北京大学学报(自然科学版) 第59卷 第5期 2023年9月
Acta Scientiarum Naturalium Universitatis Pekinensis, Vol. 59, No. 5 (Sept. 2023)
广东省基础与应用基础研究基金(2021A1515010814)和南方海洋科学与工程广东省实验室(珠海)创新团队建设项目(311021006)资助
doi: 10.13209/j.0479-8023.2023.054
收稿日期: 2022–10–16;
修回日期: 2023–02–14
蛭态轮虫广布种污轮虫(Rotaria sordida)的遗传多样性和系统发育关系
王庆† 汪文博 李莹 杨宇峰
暨南大学生态学系, 人与自然生命共同体重点实验室, 南方海洋科学与工程广东省实验室(珠海), 广州 510632; †E-mail: wq2010@jnu.edu.cn
摘要 为了解蛭态轮虫广布种污轮虫(Rotaria sordida)在中国和全球的系统发育关系, 明确蛭态轮虫遗传分化的驱动因素, 采集中国 10 省 12 市的污轮虫样品, 获得 COⅠ基因有效序列 32 条, 同时下载 GenBank 数据库中来自欧洲和大洋洲污轮虫的 47 条序列, 分别构建中国和全球两个地理尺度的贝叶斯系统发育树。结果显示, 中国污轮虫 COⅠ序列分为两个分类簇, 其系统发育关系不仅受到地理位置的影响, 还与生境类型有关; 全球污轮虫 COⅠ序列由两个分类簇构成, 分类簇内部分中国序列与欧洲和大洋洲序列形成同一分支。生境类型相同的污轮虫具有较高的同源性, 推测污轮虫系统发育受到地理分布和生境异质性的共同驱动。基于广义混合 Yule 溯祖模型的遗传多样性分析结果表明, 中国污轮虫 COⅠ序列鉴定出 22 个单倍型和 12 个分子隐种; 全球污轮虫 COⅠ序列鉴定出 45 个单倍型和 28 个分子隐种, 证明污轮虫形态学物种内具有较高的隐藏多样性, 推测生境异质性对隐种的发生具有重要作用。研究结果初步阐述了中国及全球污轮虫的系统发育关系和遗传多样性, 补充了中国陆生生境蛭态轮虫的 COⅠ基因数据库, 可为中国和世界谱系的生物地理学研究提供基础数据。
关键词 蛭态轮虫; 系统发育; 生物地理; 遗传多样性; COⅠ基因
作为典型的微型浮游生物, 轮虫存在大量的亚种和变种[1]。蛭态轮虫因其无性生殖、低湿休眠和体型微小易于扩散等特点, 一直是研究遗传进化和谱系生物地理学的模式物种[2]。随着 DNA 条形码技术的发展, 蛭态轮虫同一形态学物种内大量分子隐种(cryptic species)和隐藏多样性(hidden diversity)被发现[3]。
全球目前共记录蛭态轮虫 500 余种, 已证实其中地理分布范围广的广布种往往包含较高的隐藏多样性, 如转轮虫(Rotaria rotatoria)和游荡盘网轮虫(Adineta vaga)等广布种的隐种高达 30 余种[4–5]。丰富遗传多样性的发生与蛭态轮虫独特的生物学特征有关。蛭态轮虫微小的体型使其能够通过气流快速传播到全球各个角落, 其分布具有极大的时空异质性。在生态方面, 蛭态轮虫可适应不同的微生境(苔藓、落叶、土壤和水体等), 占据着不同的生态位, 生境异质性在一定程度上影响了轮虫的群落结构[6]。通常, 生物的共存或同域分化需要产生生殖隔离, 而蛭态轮虫属于严格的孤雌生殖, 在进化过程中较易积累更多的变异, 从而导致隐种发生, 有利于遗传分化和新物种的产生[7–8]。另外, 蛭态轮虫在遭遇不利环境条件(如低温和干燥等)时, 会产生休眠体, 这些休眠体可以长时间保存, 待环境条件适宜时萌发, 在新的栖息环境中迅速恢复生命活动, 重新建立种群[2,9–10]。这种适应不定期干燥的能力诱导蛭态轮虫 DNA 双链断裂, 并在修复过程中捕获外源基因[11]。研究表明, 在蛭态轮虫基因组中约有 10%的活跃基因是外源的[12], 其中 80%的外源基因编码酶蛋白在蛭态轮虫生理生化功能中具有重要贡献, 这一特点称为水平基因转移, 在遗传进化中起到至关重要的作用[13–14]。
污轮虫(Rotaria sordida)是旋轮科(Philodinidae)轮虫属(Rotaria)的一种蛭态轮虫, 可作为研究系统发育和生物地理学的模式物种。首先, 污轮虫形态特征鲜明, 爬行态时即可鉴定, 无需较强的分类学基础也可识别该轮虫。其鉴定特征为喙(Proboscis)和刺戟(Spur)长, 躯干部皮肤粘附大量杂质, 呈黄褐色, 不透明, 无眼点; 而大部分蛭态轮虫的种类鉴定依据摄食态时才可观察到的轮盘大小、上唇片形状等细节特征, 鉴定难度大, 耗时长, 无法快速准确地获得大量用于研究的个体。其次, 污轮虫栖息在苔藓、落叶、土壤和水体等不同水陆生境中, 具有广泛的环境适应性[15–16]; 而其他种类轮虫, 特别是常用于遗传分化模型的单巢纲轮虫, 多数仅生活在水体等单一生境中, 无法考量生境异质性和生态位差异对其遗传多样性的影响。同时, 污轮虫在世界范围内普遍分布[1], 甚至环境条件恶劣的南极大陆也有报道[17], 便于在全球尺度上说明蛭态轮虫的生物地理分布格局和系统发育情况。目前已有研究表明, 污轮虫是一种包含较多分子隐种的种复合体[4], 但对其遗传分化的驱动因素和与生境类型之间的联系关注较少[4], 且其遗传多样性数据库中尚缺乏中国区域的信息。
中国国土面积大, 气候和地形复杂多样, 具有多个生物多样性热点地区, 但与欧洲国家相比, 中国的蛭态轮虫研究仍处于起步阶段, 主要关注其物种多样性和生态毒理学, 尚无有关蛭态轮虫系统发育和谱系生物地理学方面的研究[15–16,18–19], 中国蛭态轮虫遗传数据库亟待补充。因此, 本研究以易识别、易获取、环境适应性强、分布范围广的污轮虫为研究对象, 获取中国 10 省两类生境(苔藓和落叶)广布种污轮虫的线粒体细胞色素 C 氧化酶亚基 I (cytochrome C oxidase subunit I)基因序列, 并结合全球已有污轮虫 COⅠ基因序列, 在中国和全球尺度上分析污轮虫的系统发育关系, 明确中国及全球污轮虫的生物地理分布模式和遗传多样性情况, 探究驱动其遗传分化的影响因素, 为全球蛭态轮虫种群遗传学研究提供基础资料。
1 材料与方法
1.1 采样点设置与样点信息
本研究采样范围涵盖中国 10 省 12 市, 最北部为山东泰安, 最南部为广东珠海, 纬向距离 1556.57km; 最东和最西部分别为浙江舟山和云南昆明, 经向距离 1945.86km。生境包含苔藓和落叶两类, 分别代表污轮虫的两种不同栖息生境。采样点分布地理信息如表 1 所示。
1.2 污轮虫COⅠ基因提取过程
使用毛细管挑取每一样品中的污轮虫若干只, 置于载玻片上, 使用超纯水清洗 3~5 次, 保证虫体周围和表面均无杂质颗粒, 然后将单只轮虫转移到0.2mL PCR 管中, 确保 PCR 管内只有一只轮虫, 防止实验中发生交叉污染, 以致生物地理分布结果不准确。在解剖显微镜下用解剖针将 PCR 管中的轮虫破碎, 保证 DNA 提取效果。
轮虫 DNA 提取遵循 Hotshot 方法[20–21], 配制碱性裂解液(NaOH 25 mmol/L, EDTA 0.2 mmol/L, pH 8.0)和中和缓冲液(Tris-HCl 40mmol/L, pH 5.0)。首先, 向 PCR 管中加入裂解液 10μL, 涡旋振荡使其充分混合, 放入 PCR 仪 95℃裂解 30 分钟, 结束后放入−20℃冰箱内冷冻, 5~10 分钟后将冷却的样品取出, 并加入 10μL 中和液。获得的 DNA 在−20℃环境中保存。
1.3 PCR扩增过程
PCR 反应采用 25μL 体系, 其中 DNA 模板 5μL, 实验时设置体系相同的 3 个平行样品。PCR 引物使用 LCO1490: GGTCAACAAATCATAAAGATATTGG和 HCO2198: TAAACTTCAGCCTGACCAAAAAAT CA。PCR 反应过程如下: 1)95℃预变性 1 分钟; 2)95℃变性 40 秒, 48℃退火 30 秒, 72℃延伸 45 秒, 共计 30个循环; 3)72℃延伸 5 分钟, 4℃保存[3]。1%凝胶电泳检测 PCR 扩增产物质量。PCR 产物合格后送至上海美吉生物医药有限公司, 使用 Sanger 法进行双向测序, 测序引物为 M13F(TGTAAAACGACG GCCAGT)和 M13R(CAGGAAACAGCTATGAC)。所获得的正反向两条序列使用 Bioedit 软件校对碱基, 通过 SeqMan 进行正反向序列拼接, 剪去引物片段, 得到一条完整准确的序列。
1.4 数据收集与处理
除本实验测序结果外, 从 GenBank 中下载所有上传的污轮虫 COⅠ基因序列, 共计 47 条, 平均序列长度为 600bp 左右, 这些序列对应的物种均来自其他国家。用于分析的序列GenBank登记号如下。
DQ656852.1~DQ656855.1; DQ656873.1~DQ656875.1; EF173268.1~EF173269.1; EU076863.1~EU076870.1
EU076899.1~EU076900.1; EU751058.1~EU751071.1
EU751106.1~EU751112.1; EU751119.1~EU751121.1; EU751161.1~EU751162.1; FJ426556.1~426560.1; KM0432 10.1
将全部序列通过 MAFFT 进行多重序列比对后导出, 分别在中国和全球两个地理尺度使用 BEAST构建基于贝叶斯法的系统发育进化树。使用 DNA-sp6 输出序列单倍型, 使用 R 语言 rncl 和 splits 包中的广义混合 Yule 溯祖(GMYC)模型对序列进行物种划分。
1.5 污轮虫生物地理学和生态学分析
评估该物种中的隐藏分类单元在空间上的分布是否有限, 以及它们的生境类型是否有限。在地理方面, 以中国不同的省份为单位; 在生态方面, 样品全部来源于陆生生境, 分为落叶和苔藓两大类。为了避免随机化处理中的偏差, 如果同一实体中有更多的单倍型来自同一样本, 将通过分析随机的每个样品中包含每个单倍型的一个代表来运行模拟。通过这种方式, 可以控制属于相同地理位置和生态环境的样本[3]。
表1 采样点的地理信息及样品类型
Table 1 Geographic information of sampling sites
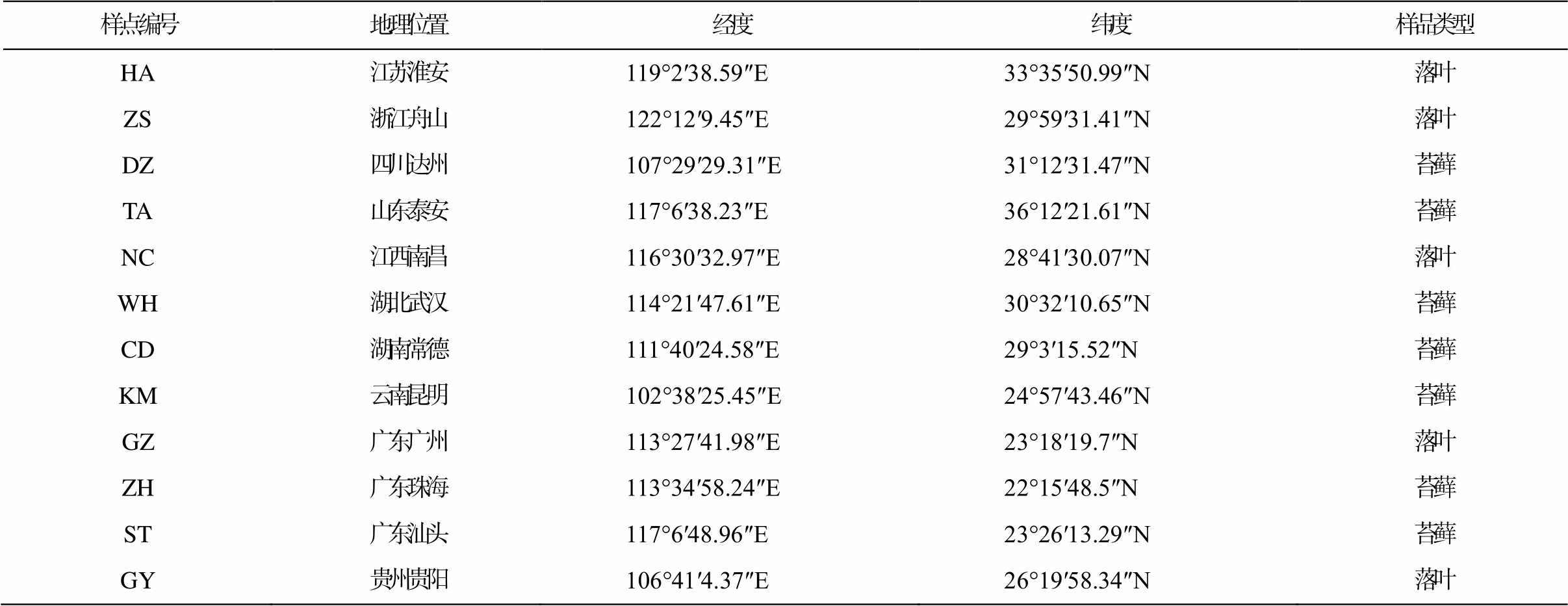
样点编号地理位置经度纬度样品类型 HA江苏淮安119°2′38.59″E33°35′50.99″N落叶 ZS浙江舟山122°12′9.45″E29°59′31.41″N落叶 DZ四川达州107°29′29.31″E31°12′31.47″N苔藓 TA山东泰安117°6′38.23″E36°12′21.61″N苔藓 NC江西南昌116°30′32.97″E28°41′30.07″N落叶 WH湖北武汉114°21′47.61″E30°32′10.65″N苔藓 CD湖南常德111°40′24.58″E29°3′15.52″N苔藓 KM云南昆明102°38′25.45″E24°57′43.46″N苔藓 GZ广东广州113°27′41.98″E23°18′19.7″N落叶 ZH广东珠海113°34′58.24″E22°15′48.5″N苔藓 ST广东汕头117°6′48.96″E23°26′13.29″N苔藓 GY贵州贵阳106°41′4.37″E26°19′58.34″N落叶
2 结果
2.1 污轮虫COⅠ基因序列分析
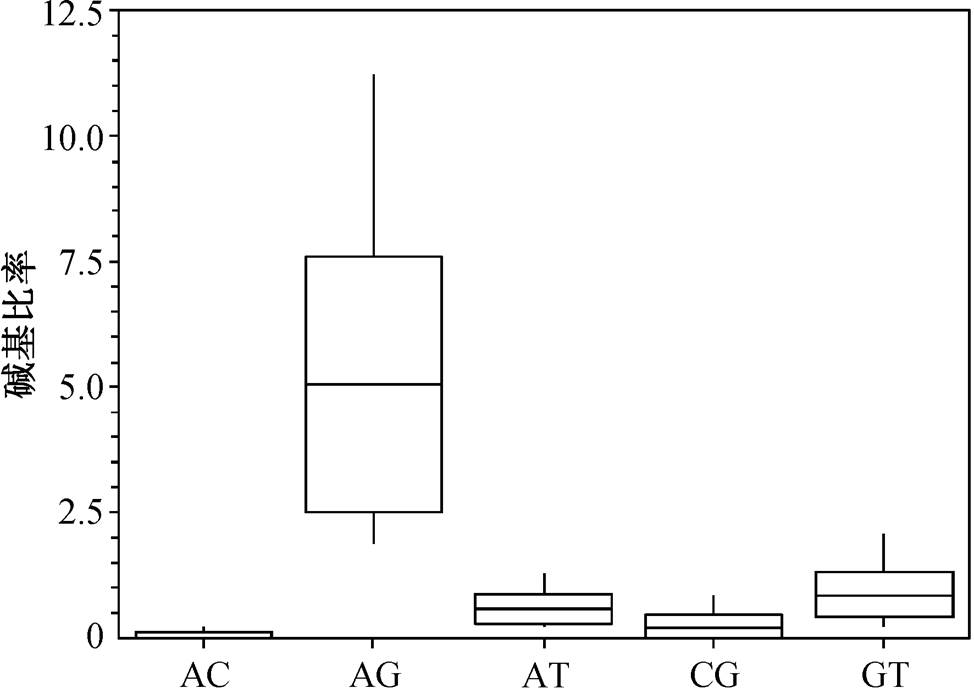
对广布种污轮虫 COⅠ基因序列进行分析, 碱基 AG 的比率较高, 为 5.0643; AC 比率较低, 仅为0.0323 (表 2, 图 1)。碱基 AG 比率的平均值与中值的差距较大, 说明样本中数值较分散。有效样本含量(effective sample size, ESS)均大于 100, 其中 CG碱基比率的有效样本含量高达 722, 说明所分析的基因序列可以进行广布种的进化时间分析。
2.2 中国各地区R. sordida遗传距离分析
根据 Kimura 2-parameter 模型计算广布种污轮虫的遗传距离, 平均遗传距离为 0.1091, 其中广东珠海与其他 11 个城市的遗传距离均大于 0.1(表3)。珠海位于热带地区, 这种基因间的高差异性可能与地理区域有关。最小种内遗传距离为 0.0099, 来自贵州贵阳和江苏淮安两个地区的序列, 其基因重组率<1%, 遗传差异较小。其次, 江西南昌、广东广州与浙江舟山污轮虫的遗传距离也较低, 分别为 0.0150 和 0.0124。最大的种内遗传距离为 0.1680, 来自湖南常德和山东泰安的污轮虫, 其基因交换概率较大, 重组率也较高。
表2 基因序列碱基比率分析
Table 2 Base ratio analysis of gene sequence
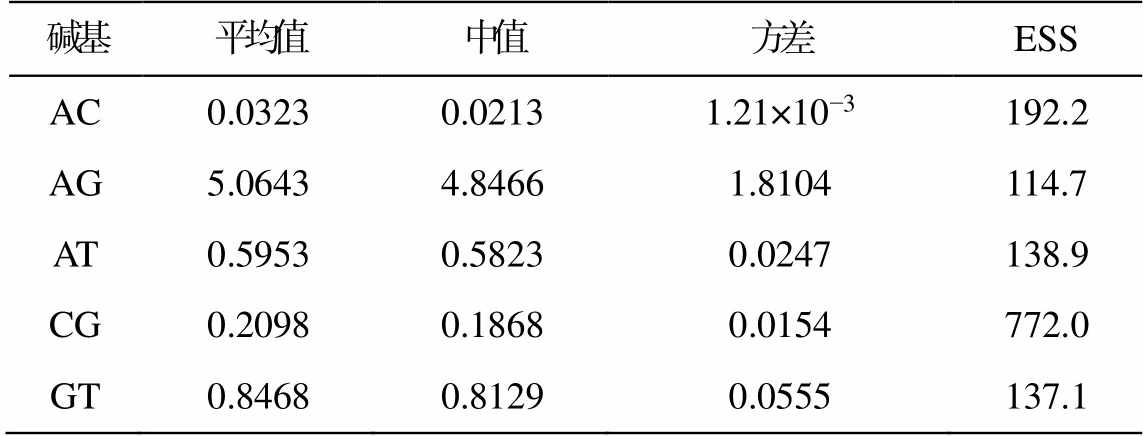
碱基平均值中值方差ESS AC0.03230.02131.21×10−3192.2 AG5.06434.84661.8104114.7 AT0.59530.58230.0247138.9 CG0.20980.18680.0154772.0 GT0.84680.81290.0555137.1
2.3 污轮虫中国地理分布格局
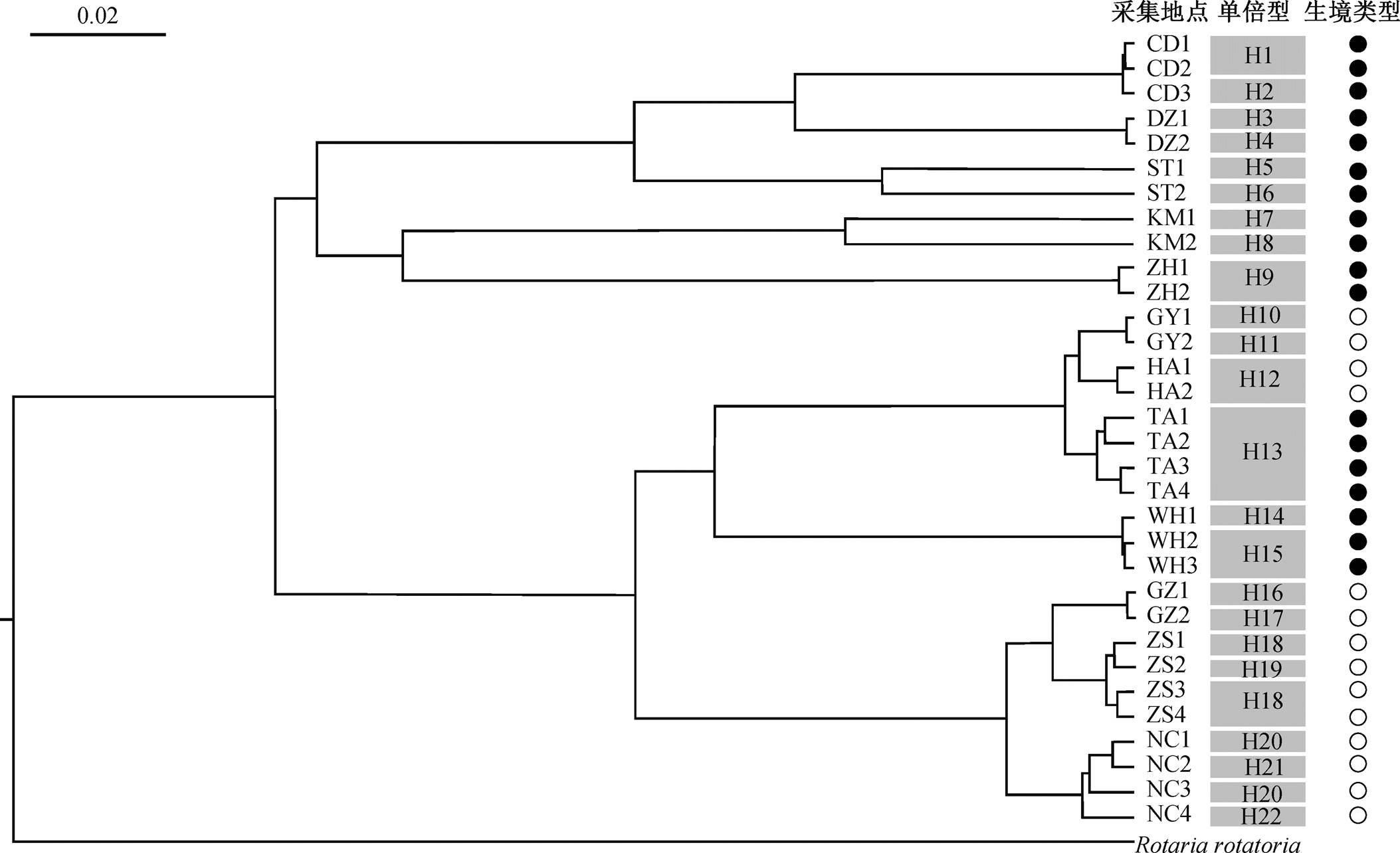
本研究获得中国 10 省 12 市的污轮虫 COⅠ基因序列 32 条, 鉴定出单倍型 22 个, 以同为轮虫属的转轮虫作为外类群, 构建基于贝叶斯分析的系统发育进化树。研究结果显示, 外类群物种转轮虫独立于污轮虫群体, 单独形成一支, 而中国不同地区来源的污轮虫聚集为一支(图 2), 验证了形态学鉴定的结果。
表3 中国不同地区污轮虫的遗传距离
Table 3 Genetic distance of R. sordida from different sites in China
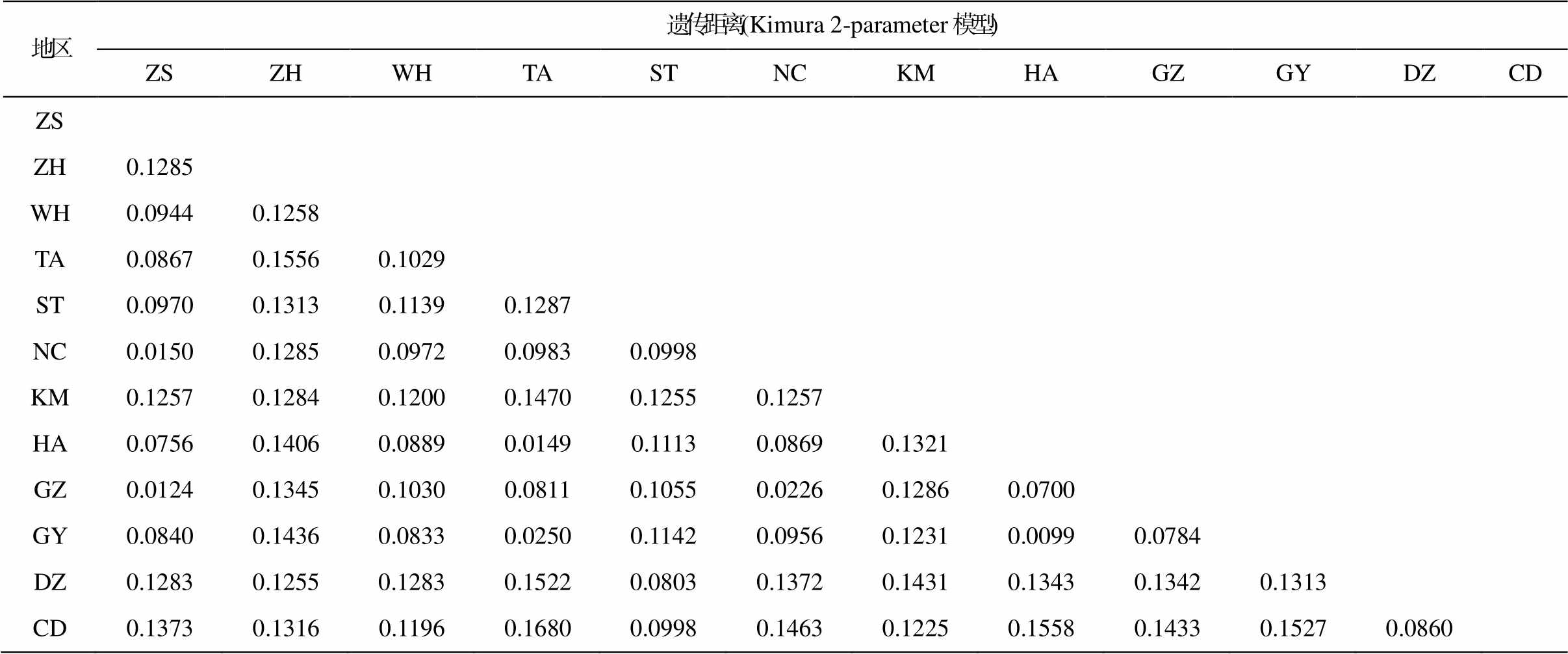
地区遗传距离(Kimura 2-parameter模型) ZSZHWHTASTNCKMHAGZGYDZCD ZS ZH0.1285 WH0.09440.1258 TA0.08670.15560.1029 ST0.09700.13130.11390.1287 NC0.01500.12850.09720.09830.0998 KM0.12570.12840.12000.14700.12550.1257 HA0.07560.14060.08890.01490.11130.08690.1321 GZ0.01240.13450.10300.08110.10550.02260.12860.0700 GY0.08400.14360.08330.02500.11420.09560.12310.00990.0784 DZ0.12830.12550.12830.15220.08030.13720.14310.13430.13420.1313 CD0.13730.13160.11960.16800.09980.14630.12250.15580.14330.15270.0860
中国污轮虫谱系内部形成两大差异明显的分类簇(图 2), 其中一大分类簇包括 NC(江西南昌)、ZS(浙江舟山)、GZ(广东广州)、WH(湖北武汉)、TA (山东泰安)、HA(江苏淮安)以及 GY(贵州贵阳) 7 省7 地的样品。该分类簇中又细分为两个明显不同的分支。南昌、舟山和广州的 COⅠ基因序列具有更高的同源性, 亲缘关系较近, 这 3 个地区的序列均来自落叶生境。另外, 泰安、淮安、贵阳和武汉四地聚为一支, 其中泰安、淮安和武汉三地的地理位置相对较近, 但淮安与另外两地的样品类型不同, 聚类依据可能更多地取决于地理因素; 贵阳位于中国西南部, 与淮安聚为一支, 其样品类型均为落叶; 同样, 同属苔藓生境的武汉和泰安的轮虫 COⅠ基因序列也因生境类型相同而聚为一支。另一分类簇包括来自 ZH(广东珠海)、KM(云南昆明)、ST(广东汕头)、DZ(四川达州)和 CD(湖南常德)的污轮虫样品, 涵盖 4 省 5 市。上述五地分属中国西南以及华南地区, 地理距离相对较近, 样品类型均为苔藓。相似的气候条件、毗邻的地理位置和相同的生境, 使得这些地区的广布种污轮虫亲缘关系较近。与其他 10 个地区相比, 昆明(KM)和汕头(ST)两地序列遗传距离较大, 但也单独聚为一支。因此, 每个地区物种基因序列的同源性是准确可靠的。
中国共有污轮虫单倍型 22 个, 其分布主要受地理位置影响, 现有分析结果中, 即使同属于一类生境, 也没发现一个单倍型在两地及以上地区存在。同一地区的污轮虫序列也可能分属于不同的单倍型, 存在同域物种分化的趋势。广东珠海、江苏淮安和山东泰安三地只有一个单倍型, 证明了这些地区污轮虫遗传上的保守性。
2.4 污轮虫全球地理分布格局
在GenBank中获取的 47 条 COⅠ基因序列来自意大利、英国、瑞士、澳大利亚、西班牙和法国等国家, 涉及欧洲和大洋洲两大区域, 生境类型包括苔藓和地衣等。污轮虫在上述国家均有分布, 表现出广泛的全球地理分布特征。
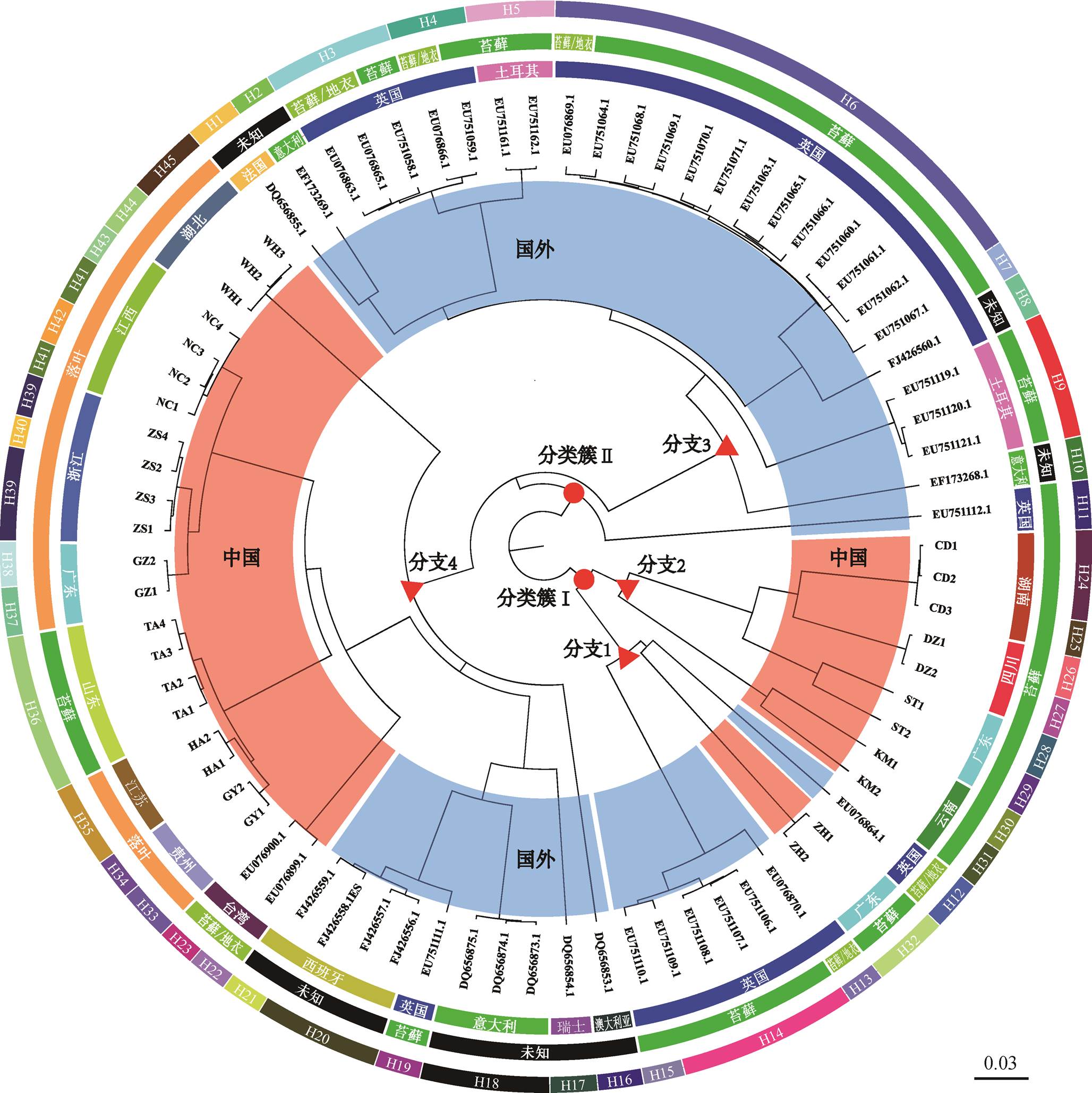
贝叶斯系统发育分析结果显示, 全球污轮虫形成两个分类簇, 均包含中国和国外两部分序列。这些国外样本的采集地点距离中国较远, 分属不同的生物地理区域, 但来自不同国家和地区的序列也可聚为亲缘关系较近的一支(图 3)。如分类簇Ⅰ分支 1中来自英国的部分序列(EU751106~EU751110, EU 076864, EU076870)与中国的珠海(ZH)基因序列聚为一支, 分支 2 中常德(CD)、汕头(ST)、达州(DZ)和昆明(KM)聚为一支。分类簇Ⅱ中按地理位置分为 3 和 4 两个分支, 分支 3 中英国样本(EU751064~ EU751081 等)与法国(DQ656855)、土耳其(EU 751161~EU751162 等)和意大利(EF173268~EF 173269)则聚为一个分支(图 3 右上), 与中国污轮虫基因序列明显分离。分支 4 中, 中国的贵阳(GY)、淮安(HA)和泰安(TA)等地区与西班牙(FJ426556~ FJ426559)、英国(EU751111)、意大利(DQ656873~ DQ656875)、瑞士(DQ656854)的序列聚集为较大的一个分支。在生境类型方面, 分类簇Ⅰ均为苔藓或地衣生境; 分类簇Ⅱ中, 除未知生境外, 分支 3 均为苔藓或地衣生境, 分支 4 包括苔藓和落叶生境, 其内部大体上按生境类型聚类。
全球污轮虫单倍型多样性高, 共鉴定出 45 个, 其多样性分布同样主要受地理位置影响, 所有单倍型仅限于一个地点, 但同一单倍型却可在不同生境中出现, 如单倍型 H3, H4 和 H6。中国污轮虫单倍型数量高于国外(图 3), 这种差异可能是由采样点的设置和采样方法的差异导致。
综上所述, 中国污轮虫基因序列分为两部分, 并与欧洲和大洋洲国家序列聚集在一起, 形成亲缘关系较近的分支。因此, 除地理隔离外, 栖息地生境对污轮虫的物种演化、生存和传播同样有重要的影响。
2.5 基于DNA分类的隐藏多样性
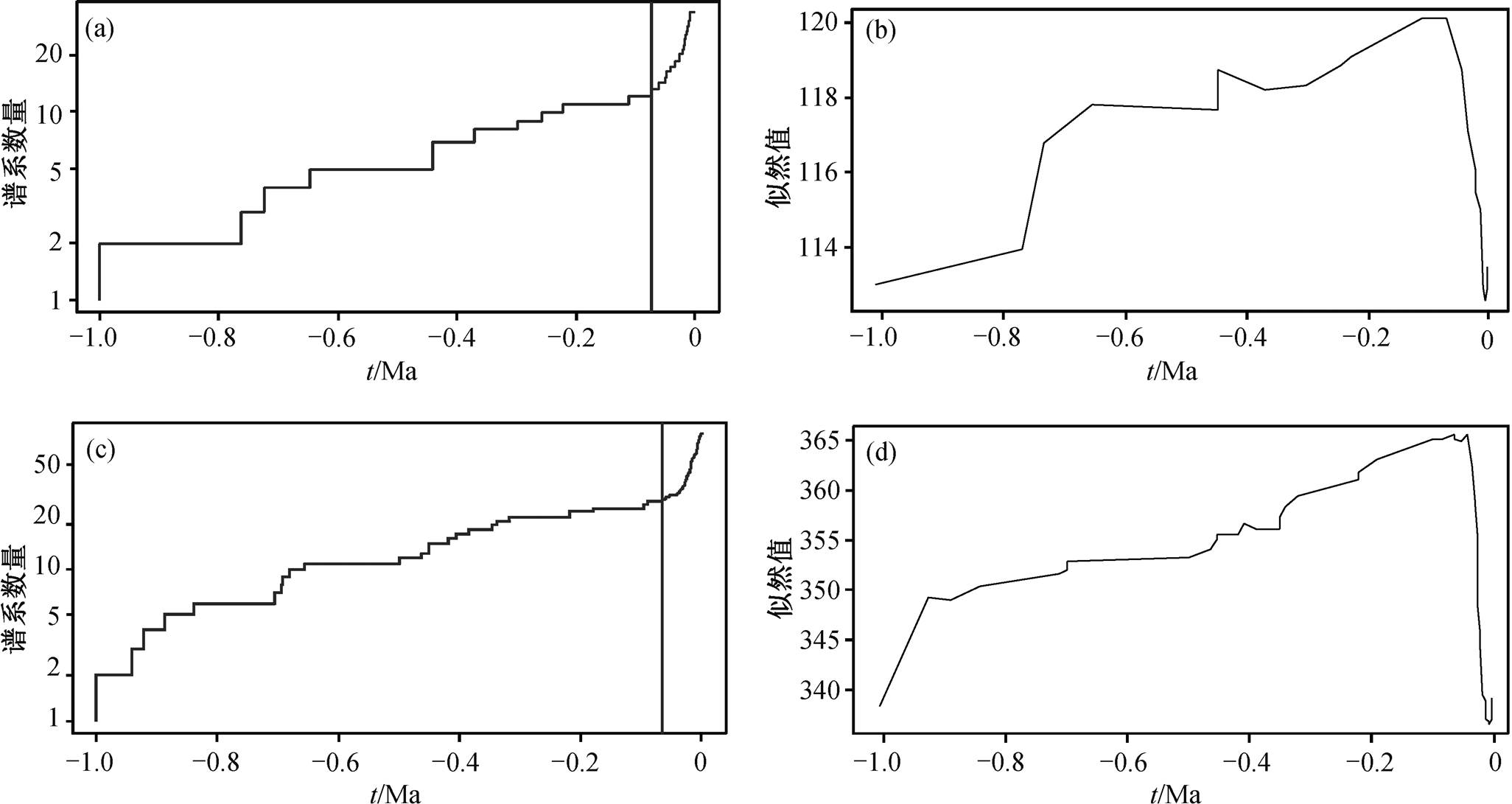
分子隐种指存在于同一形态学物种下, 形态学无法区分, 但遗传信息存在差异的物种, 体现物种的遗传多样性和发生趋势。图 4(a)和(c)中的曲线表示枝长速率变化的趋势, 枝长速率呈指数形式急剧上升时, 横坐标转折点对应的谱系数值就是对应物种界定的数量, 此时横坐标代表种(Yule模型)–种群的转折发生时间。中国地区分布的污轮虫种群的转折点发生在 0.0719 百万年前, 此时 Y 轴对应的界定物种数量为 7(图 4 和表 4)。图 4(b)反映似然值随时间的变化, 其中似然比为 19.96991(p=0.000925706 <0.001), 似然比结果极显著(表 4)。
目前全球已获取的全部污轮虫 COⅠ基因序列中, 种群的转折发生点在 0.0671 百万年前, 此时相对应的物种界定数量为 15(不包括单个体数)(图4(c), 表 4), 似然比为 53.93846(p=1.938227×10−12 <0.001), 似然比结果也极其显著(图 4(d), 表 4)。
从上述分析可知, 通过污轮虫的 DNA 分类方法, 发现该物种具有较高的隐藏多样性。其中, 在所研究的中国各省市中, 界定物种数量包含单个体数(隐藏实体 entities)为 6~14 个, 全球范围内该物种的隐藏实体范围是 25~31 个, 进一步证明基于分子鉴定的隐藏多样性在蛭态轮虫类群中普遍存在。在不同栖息地的广布性物种中, 隐藏分类单元的数量更高时, 我们假设大多数广布物种将具有较高的隐藏多样性。
表4 基于GMYC模型的物种界定
Table 4 Species delimitation based on GMYC model
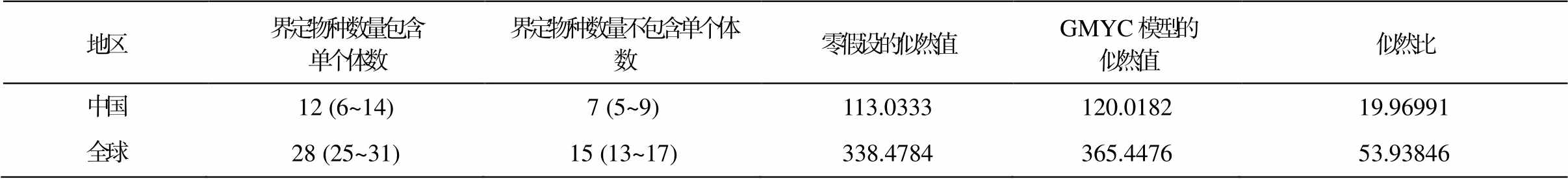
地区界定物种数量包含单个体数界定物种数量不包含单个体数零假设的似然值GMYC模型的似然值似然比 中国12 (6~14)7 (5~9)113.0333120.018219.96991 全球28 (25~31)15 (13~17)338.4784365.447653.93846
3 讨论
本研究发现污轮虫具有较高的隐藏多样性, 结果显示中国污轮虫的分子隐种数量为 12 种, 全球范围的分子隐种数高达 28 种。这种高隐藏多样性现象在轮虫这类微型生物中普遍存在[22–23]。例如, 单巢纲褶皱臂尾轮虫(Brachionus plicatilis)的隐种数为22 种, 是目前记录的单巢纲轮虫隐藏多样性最高的物种[23]; 蛭态轮虫中转轮属的转轮虫的隐种数达 34种[4], 盘网轮属的游荡盘网轮虫多达 36 种[5]。本研究中, 全球范围的污轮虫隐种数量也高于单巢纲轮虫, 可能是因为它们独特的孤雌生殖特性[4,10]。污轮虫广泛分布在苔藓、落叶和水生生境中, 这种宽生态位也促进了隐种的分化, 即隐种的发生常常出现在可兼性生活在不同生境的物种[22,24], 而对于那些只能生存在单一生境中的种类, 即使扩大 COⅠ基因数据的数量及地理尺度, 其隐藏实体的个数可能仍为 1, 即未发生隐种分化[4]。例如, 生态位狭窄的轮虫属 R. magnacalcarata 和 R. socialis 分别附生在水虱腹部和腿部, 它们的隐种数量为 2 和 1, 而生境广适性物种如四角粗颈轮虫(Macrotrachela quad-ricornifera)、转轮虫和污轮虫具有更高的隐藏多样性, 这也间接地反映了生境异质性对物种隐种发生的影响[4]。
污轮虫广泛分布于欧洲、亚洲多个国家的多种生境中, 即使在环境条件恶劣的南极洲, 污轮虫也依然可以生存[17,25]。其休眠体在全世界散布的可能性非常高, 与其他蛭态轮虫广布种一样, 污轮虫在全世界广泛分布, 本地物种的丰富度可以反映该物种的全球多样性[26]。Fontaneto 等[4]通过调查几种广布蛭态轮虫, 发现地理隔离是推动广适种隐藏多样性变异的主要因素。本文研究结果表明, 中国分布的广布种污轮虫的系统发育受到地理区域和生境类型两大因素影响, 无论从大的分类簇还是小的分支来看, 其赖以生存的不同类型微生境对污轮虫的系统发育均有影响。同样, 游荡盘网轮虫的遗传多样性也受到生境类型影响, 但与地理分布无关[5]。对于蛭态轮虫这类微型动物, 占有极小面积的生存环境就具有丰富的物种多样性[10,26], 其休眠状态下体型小, 抗逆性强, 易通过气流或动物携带引起种群的大规模扩散[10,26]。所以短时间内, 地理位置相近可以作为解释蛭态轮虫同源性的依据。然而, 生境类型相同的蛭态轮虫种群面临的生存压力相近, 易受到环境过滤效应影响, 即在扩散传播充分的条件下, 生态位与环境相适宜的物种得以定殖[5]。这类传统物种的隐种形态上存在细微差别, 是物种适应生境而产生形态可塑性的体现[5]。另外, Jaturapru-ek 等[27]通过调查泰国 390 个水体的轮虫属蛭态轮虫发现, 生境类型及不同生境间固有的生态差异是造成蛭态轮虫群落差异的最主要因素。Liang 等[28]在研究中国和美国多肢轮虫(Polyarthra dolichoptera和 P. vulgaris)的遗传距离与地理距离关系时, 发现中美大陆多肢轮虫基因流受到生境异质性、长距离定殖和海洋屏障混合效应影响。结合对污轮虫谱系生物地理学的研究结果, 发现对于蛭态轮虫这类微型动物, 其遗传多样性和系统发育关系不仅受地理距离的影响, 特定生存环境(生境)也是隐种发生的重要驱动因素[5,28]。在未来的研究中, 应探索隐种遗传信息、形态学和生境之间的关系, 以便加强对生境驱动蛭态轮虫遗传分化机制的理解。另外, 本研究中分类簇Ⅰ包含中英同属苔藓或地衣生境的污轮虫序列, 证明相同生境物种有着更高的系统发育同源性, 但与分支 3 中同属英国苔藓生境的明显分化暗示除地理和生境因素外, 还有其他变量导致分化差异的出现, 需要获取更广泛地域、多样化生境以及多种物种的遗传信息来进一步探索。
中国地域南北跨度大, 地形气候复杂多样, 但目前有关蛭态轮虫的多样性和谱系生物地理学研究仍然较少[15–16], 一些具有代表性的特殊生境(如青藏高原、广西喀斯特等)值得调查, 以便获取蛭态轮虫的形态学和遗传多样性数据, 这对于理解蛭态轮虫全球系统发育关系和生物地理学模式具有重要意义。本研究中的 COⅠ序列局限于中国部分省市, Genbank 数据库中污轮虫数据多集中在欧洲和大洋洲等地, 尚缺乏世界其他大洲的序列数据。如果能扩大采样范围, 增加样本数量, 探索更多微生境(如露水等[15]), 特别是对蛭态轮虫研究较少的热带地区, 将对研究全球蛭态轮虫谱系生物地理学和遗传分化起到重要作用。另外, 全球关于蛭态轮虫系统发生学的研究大多只关注单一生境(如苔藓), 缺乏对落叶等不同生境中生存物种遗传多样性的比较研 究[3,16–17,29]。同时, 由于单巢纲轮虫和缓步类在形态或生物学特征方面与蛭态轮虫具有相似性, 加强这些物种遗传多样性的比较研究, 对揭示微型动物进化和变异驱动因素具有重要意义。
4 结论
本研究采集中国 10 省 12 市的污轮虫样品, 成功地获取该物种的 COⅠ基因序列 32 条, 从 Gen-bank 下载欧洲和大洋洲污轮虫 COⅠ序列 47 条, 在中国和全球两个地理尺度上分析污轮虫的系统发育状况。本研究发现污轮虫的系统发育关系不仅受地理距离影响, 而且生境类型相同的污轮虫具有更高的同源性。基于 GMYC 模型, 中国共鉴定出污轮虫分子隐种 12 个, 全球共发现污轮虫分子隐种 28 个, 证明污轮虫具有较高的隐藏多样性, 推测隐种发生的原因可能是受生境异质性和地理隔离的共同作用。另外, 本文获取的广布种污轮虫 COⅠ序列极大地丰富了中国蛭态轮虫, 特别是陆生种类的遗传多样性信息库。未来的研究中, 可使用不同分子标记获取更多不同生境不同蛭态轮虫物种的遗传信息, 为构建中国蛭态轮虫遗传信息库, 强化蛭态轮虫分子鉴定方法, 深入研究蛭态轮虫谱系生物地理学和遗传进化奠定基础。
参考文献
[1] Segers H. Annotated checklist of the rotifers (Phylum Rotifera), with notes on nomenclature, taxonomy and distribution. Zootaxa, 2007, 1564(1): 1–104
[2] Shmakova L, Malavin S, Iakovenko N, et al. A living bdelloid rotifer from 24,000-year-old Arctic perma-frost. Current Biology, 2021, 31(11): R712–R713
[3] Fontaneto D, Eckert E M, Aninic N, et al. We are ready for faunistic surveys of bdelloid rotifers through DNA barcoding: the example of Sphagnum bogs of the Swiss Jura Mountains. Limnetica, 2019, 38(1): 213–225
[4] Fontaneto D, Kaya M, Herniou E A, et al. Extreme levels of hidden diversity in microscopic animals (Rotifera) revealed by DNA taxonomy. Molecular Phy-logenetics and Evolution, 2009, 53(1): 182–189
[5] Fontaneto D, Iakovenko N, Eyres I, et al. Cryptic diversity in the genus Adineta Hudson & Gosse, 1886 (Rotifera: Bdelloidea: Adinetidae): a DNA taxonomy approach. Hydrobiologia, 2011, 662(1): 27–33
[6] Kaya M, Erdoğan S. Testing the habitat selectivity of bdelloid rotifers in a restricted area. Turkish Journal of Zoology, 2015, 39(6): 1132–1141
[7] Barraclough T G, Birky C W, Burt A. Diversification in sexual and asexual organisms. Evolution, 2003, 57 (9): 2166–2172
[8] Fontaneto D, Barraclough T G. Do species exist in asexuals? Theory and evidence from bdelloid rotifers. Integrative and Comparative Biology, 2015, 55(2): 253–263
[9] Bohonak A J, Jenkins D G. Ecological and evolu-tionary significance of dispersal by freshwater inverte-brates. Ecology Letters, 2003, 6(8): 783–796
[10] Ricci C. Bdelloid rotifers: ‘sleeping beauties’ and ‘evo-lutionary scandals’, but not only. Hydrobiologia, 2017, 796(1): 277–285
[11] Hespeels B, Knapen M, Hanot-Mambres D, et al. Gateway to genetic exchange? DNA double-strand breaks in the bdelloid rotifer Adineta vaga submitted to desiccation. Journal of Evolutionary Biology, 2014, 27(7): 1334–1345
[12] Boschetti C, Carr A, Crisp A, et al. Biochemical diver-sification through foreign gene expression in bdelloid rotifers. PLoS Genetics, 2012, 8(11): e1003035
[13] Welch D B M, Ricci C, Meselson M. Bdelloid rotifers: progress in understanding the success of an evolu-tionary scandal // Schön I, Martens K, Dijk P. Lost sex. Dordrecht: Springer, 2009: 259–279
[14] Debortoli N, Li Xiang, Eyres I, et al. Genetic exchange among bdelloid rotifers is more likely due to horizontal gene transfer than to meiotic sex. Current Biology, 2016, 26(6): 723–732
[15] Zeng Yue, Wei Nan, Wang Qing, et al. Bdelloid rotifers (Rotifera, Bdelloidea) of China: diversity and new records. Zookeys, 2020, 941: 1–23
[16] 汪文博, 王庆, 李莹, 等. 落叶生境蛭态轮虫物种多样性及四种中国新记录种. 水生生物学报, 2021, 45(2): 436–445
[17] Cakil Z V, Garlasché G, Iakovenko N, et al. Compa-rative phylogeography reveals consistently shallow genetic diversity in a mitochondrial marker in Antarc-tic bdelloid rotifers. Journal of Biogeography, 2021, 48(7): 1797–1809
[18] Xiang Xianling, Jiang Ruiming, Chen Yinging, et al. Regulation of heat shock protein 70 (Hsp70) levels in the bdelloid rotifer Rotaria rotatoria under tempera-ture stress. Israel Journal of Ecology and Evolution, 2017, 63(1): 69–77
[19] Zhu Lingyun, Huang Rong, Zhou Libin, et al. Res-ponses of the ecological characteristics and antioxi-dant enzyme activities in Rotaria rotatoria to UV-B radiation. Hydrobiologia, 2021, 848(20): 4749–4761
[20] Montero-Pau J, Gómez A, Muñoz J. Application of an inexpensive and high-throughput genomic DNA extra-ction method for the molecular ecology of zooplank-tonic diapausing eggs. Limnology and Oceanography: Methods, 2008, 6(6): 218–222
[21] Fontaneto D, Flot J F, Tang C Q. Guidelines for DNA taxonomy, with a focus on the meiofauna. Marine Biodiversity, 2015, 45(3): 433–451
[22] Fontaneto D, Barraclough T G, Chen K, et al. Mole-cular evidence for broad-scale distributions in bdelloid rotifers: everything is not everywhere but most things are very widespread. Molecular Ecology, 2008, 17(13): 3136–3146
[23] Gabaldón C, Fontaneto D, Carmona M J, et al. Ecological differentiation in cryptic rotifer species: what we can learn from the Brachionus plicatilis complex. Hydrobiologia, 2017, 796(1): 7–18
[24] Kaya M, Herniou E A, Barraclough T G, et al. Inconsistent estimates of diversity between traditional and DNA taxonomy in bdelloid rotifers. Organisms Diversity & Evolution, 2009, 9(1): 3–12
[25] Iakovenko N S, Smykla J, Convey P, et al. Antarctic bdelloid rotifers: diversity, endemism and evolution. Hydrobiologia, 2015, 761(1): 5–43
[26] Fontaneto D, Ficetola G F, Ambrosini R, et al. Patterns of diversity in microscopic animals: are they com-parable to those in protists or in larger animals?. Global Ecology and Biogeography, 2006, 15(2): 153–162
[27] Jaturapruek R, Fontaneto D, Maiphae S. The influence of environmental variables on bdelloid rotifers of the genus Rotaria in Thailand. Journal of Tropical Eco-logy, 2020, 36(6): 267–274
[28] Liang D, McManus G B, Wang Q, et al. Genetic diffe-rentiation and phylogeography of rotifer Polyarthra dolichoptera and P. vulgaris populations between Sou-theastern China and eastern North America: high inter-continental differences. Ecology and Evolution, 2022, 12(5): e8912
[29] 王庆, 李莹, 汪文博, 等. 广东省蛭态轮虫物种多样性及其系统发育研究. 中国环境科学, 2021, 41 (9): 4367–4377
Genetic Diversity and Phylogenetic Relationship of the Cosmopolitan Bdelloid Rotifer Rotaria sordida
WANG Qing†, WANG Wenbo, LI Ying, YANG Yufeng
Department of Ecology, Key Laboratory of Philosophy and Social Science in Guangdong Province, Jinan University, Southern Marine Science and Engineering Guangdong Laboratory (Zhuhai), Guangzhou 510632; † E-mail: wq2010@jnu.edu.cn
Abstract In order to understand the phylogeny of Rotaria sordida in China and the world, and to clarify the driving factors of the genetic differentiation of bdelloid rotifers, R. sordida samples were collected from 12 cities in 10 provinces of China, and 32 COⅠgene sequences were obtained. Moreover, 47 COⅠgene sequences from Europe and Oceania in the GenBank database were downloaded, and the Bayesian phylogenetic tree was constructed at China and global scales, respectively. The results showed that the COⅠgene sequences of R. sordida in China were divided into two taxonomic clusters, and their phylogenetic relationships were not only affected by geographical location, but also related to habitat types. At global level, all sequences were also divided into two clusters, and some sequences in China were clustered with sequences from Europe and Oceania. The bdelloids with the same habitat type in the two clusters had high homology. It was speculated that the phylogeny of bdelloid rotifers was driven by geographic distribution and habitat heterogeneity. Genetic diversity analysis based on the Generalized Mixed Yule Coalescent model (GMYC) showed that 22 haplotypes and 12 cryptic species were identified from the COⅠsequence of R. sordida in China; 45 haplotypes and 28 cryptic species were identified from the global COⅠsequence. This indicated the high hidden diversity in R. sordida species complexes. Therefore, habitat heterogeneity played an important role in cryptic species differentiation of R. sordida. This study preliminarily explained the phylogenetic relationship and genetic diversity of R. sordida in China and the world. Furthermore, this study supplemented the COⅠgene database of bdelloid rotifers in China, and provided basic data for lineage biogeographic studies in the future.
Key words bdelloid rotifer; phylogeny; biogeography; genetic diversity; COⅠgene