 , 边的特征矩阵表示为
, 边的特征矩阵表示为 。
。北京大学学报(自然科学版) 第62卷 第2期 2026年3月
Acta Scientiarum Naturalium Universitatis Pekinensis, Vol. 62, No. 2 (Mar. 2026)
doi: 10.13209/j.0479-8023.2025.093
国家自然科学基金(62403437, 62576098)、河南省重点研发与推广专项(242102211039)和郑州轻工业大学校级青年骨干教师培养项目(13502010009)资助
收稿日期: 2025–03–05;
修回日期: 2025–08–20
摘要 针对药物和靶标等生物分子的复杂关系, 融合多源异质图和异质门图卷积, 设计一种异质生物图动态表示学习方法 HGGCN。该方法利用异质图门卷积的门控通道和特征通道, 自适应建模异质生物图中的互作用模式, 融合增强复杂网络的拓扑结构和语义信息, 实现药物与靶标的知识表示以及药物–靶标互作用信息挖掘。实验结果表明, 所提模型的性能优于现有药物–靶标互作用预测方法, 是一种精准的药物–靶标关联预测工具, 可以支持复杂生物数据建模和疾病精准治疗。
关键词 复杂生物网络; 多源异质图; 神经网络; 药物–靶标互作用
肿瘤、癌症和心血管等疾病均由遗传和环境等因素共同作用导致, 从遗传分子层面开展复杂疾病的智能计算和诊断方法研究是当前备受关注且极具挑战性的领域[1–5], 包括药物–靶标互作用、疾病非编码 RNA 互作用、药物互作用以及疾病的致病基因等方面。靶标是能被药物作用的生物分子, 包括核受体、离子通道、G 蛋白偶联受体和酶等[6–8], 药物–靶标互作用预测研究是药物重新定位和药物研发的关键步骤, 药物功能与靶标的特异性识别联系紧密。药物通常激活或抑制人体中某些蛋白质活性[3], 药物–靶标的互作用预测是药物研发的重要方向[9], 在早期药物筛选、老药新用以及探讨药物毒副作用等环节起着重要作用[10]。然而, 基于蛋白芯片以及亲和层析等生物实验[7,9,11]的鉴定方法具有精度低、成本高和周期长等局限。
图神经网络等深度计算模型可以揭示复杂网络的深层拓扑信息, 是节点分类和链接预测的重要算法[3,12–14]。然而, 真实世界中的图结构数据往往由不同类型的实体对象及其复杂关系连接构成[15–17]。在异质图结构数据中, 图数据的拓扑模式可以描述复杂的语义信息和近邻部分的结构信息[7,15,18]。但是, 在药物–靶标相互作用预测中, 深度模型在梯度反向传播的过程中容易出现梯度消失[7,13], 导致不能充分提取长距离依赖的上下文特征。针对药物和靶标等生物分子间的复杂作用及多类型的节点和边, 基于图数据结构的药物–靶标互作用预测方法面临以下挑战: 1)药物与靶标之间的关系是复杂和异质的; 2)不同生物分子的交互使得传统的网络方法无法充分建模不同类型节点之间的交互信息; 3)图神经网络的过平滑问题使得生物分子间的差异性减低。Yang 等[15]集成化学分子、基因、通路和疾病等实体信息, 构建一个大规模异质生物网络, 利用图卷积提升药物–靶标互作用预测任务性能, 获得很好的结果。此外, 基于多模态表示学习和门控线性单元机制, Peng 等[19]提出一种多模态门控网络, 用于提升药物–靶标互作用预测性能。基于多阶门控卷积与多注意力机制融合, Li 等[20]开发一种有效的深度计算模型提取药物和蛋白质特征, 用于提升药物–靶标相互作用预测模型的性能。这类门控线性单元方法提供了一种动态的特征选择和控制机制, 自动筛选药物靶标预测的判别性模式信息。
随着高通量测序和自动化技术的发展以及药物和疾病相关数据库的建立[4–5,9], 设计更加精准的高效的自动化计算方法, 大规模和高置信度地预测药物与疾病间的潜在关联, 成为药物研发领域的研究热点。目前, 药物–靶标互作用预测方法[7,9,13,18,21]可分为对接模拟方法和机器学习方法[3,6,22–24]。对接模拟方法主要包括基于配体的对接模拟方法和基于分子对接模拟的方法。但是, 基于对接模拟的预测方法比较耗时, 且在靶标蛋白的结构信息未知时无法对接。当药物和靶标的已知配体较少时, 基于配体方法的预测能力较低。基于机器学习的药物–靶标互作用预测方法主要采用支持向量机、贝叶斯、矩阵分解和深度计算等模型。药物–靶标的异质网络[25–26]中包含大量高维信息和复杂关联数据, 然而已开发的药物–靶标互作用预测方法依赖于药物和蛋白质的统计特征, 未充分考虑药物和蛋白质等生物分子的拓扑结构特征, 存在特征表示和预测能力不足的问题。
本文鉴于深度神经网络在图像处理[27]、情绪识别[28]、药物靶标[29]及基因信号分析[30]已获得很好的结果, 针对药物–靶标互作用预测方法的问题和挑战, 整合多种药物靶标关联网络, 构建异质生物图数据结构, 提出异质图中不同生物分子节点间的语义关联和长距离依赖特征的嵌入表示学习方法, 提升药物–靶标关系预测的性能。本文将门控机制引入药物–靶标异质网络的特征学习中, 设计深度异质图门卷积网络(heterogeneous graph gated convolutional networks, HGGCN), 提出一种拓扑结构及语义信息增强的异质生物图动态表示学习方法。HGGCN 综合考虑异质图中药物–靶标的局部拓扑空间语义和长期序列的组合特征, 将集成的药物和蛋白质特征以及经验证的药物互作用、药物蛋白质关联、药物疾病关联、药物副作用、蛋白质互作用和蛋白质疾病关联整合进药物–靶标的异质图中。然后, 基于图卷积的指数型门控机制, 控制聚合异质邻居顶点特征, 并将聚合后的特征与中心顶点的特征相融合, 获得药物和靶标的特征表示。最后, 设计一种铰链熵损失函数, 根据 HGGCN 预测的概率与真实标签间的损失, 对整个模型进行端到端的训练, 完成参数学习和模型的梯度更新, 将药物和靶标特征输入预测模块中, 得出药物与靶标之间存在互作用的概率, 实现药物与靶标知识表示以及药物–靶标互作用信息挖掘。
传统的计算方法主要有分子对接方法和配体方法。Lounkine 等[31]以药物结构为出发点, 对药物结构进行了定量比较, 提出一种将多种相似度计算相结合的框架。Öztürk 等[32]利用配体已知的化学结构信息、蛋白质序列信息以及类似靶蛋白之间存在的类似的配体预测潜在的药物靶标。以差分进化模型为基础, Thomsen 等[33]提出一种基于启发式搜索的分子对接算法, 引入重新排序评分函数, 从对接算法获得的结果中识别出置信度最高的对接对。Goo-dsell 等[34]结合经验自由能力场和 Lamarckian 遗传算法, 设计一种分子对接的软件 AutoDock, 基于网格的对接模型和柔性侧链技术, 预测蛋白质配体的结合位点。基于配体的方法是利用靶标序列信息和药物信息, 从已知药物–靶标对中提取信息, 然后筛选出候选分子。此外, 当靶标蛋白的三维结构不明确时, 分子对接方法的预测性能下降。Mei 等[17]引入高斯函数, 根据邻居与查询中的药物–靶标距离, 逐渐降低邻居节点的影响, 提出一种基于二部局部的邻居的交互轮廓的药物–靶标关系推断模型。Zheng 等[21]提出一种多相似度协同矩阵因子分解模型, 将药物和靶标投影到一个共同的低秩特征空间中, 与药物和靶标上的加权相似度矩阵相一致, 预测药物–靶标关系。然而, 基于传统计算的药物–靶标互作用预测方法无法处理大批量的药物组学数据, 不能建模复杂网络的拓扑结构以及高阶语义信息。
高通量测序技术产生的海量药物组学数据为研究药物–靶标互作用的智能预测提供支持。基于相似的药物趋向于靶向相似的蛋白质的原理, 可将药物–靶标互作用预测任务视为一种分类任务, 研究和设计各种深度计算方法。Peng 等[7]基于蛋白质、疾病、药物、副作用和实体间的关联信息, 提出一种异质图卷积网络预测药物–靶标互作用。Zheng 等[21]将药物和靶标的多种相似性信息转换到同一特征空间, 利用协同矩阵分解算法, 实现药物靶标关系的预测。Xia 等[35]基于药物与靶标之间的相互作用、药物–药物相似性和蛋白质–蛋白质相似性, 提出一种半监督学习方法——拉普拉斯正则化最小二乘法, 进行药物靶标互作用预测。Luo 等[18]基于重启随机游走算法, 整合多种药物和蛋白质信息, 构建异构网络来预测药物–靶标互作用。Li 等[22]利用变分自编码器, 研究双瓦瑟斯坦生成对抗网络和梯度惩罚策略, 生成边缘信息和增强特征表示, 设计一种药物–靶标相互作用预测的深度计算方法。Zhu 等[23]整合多个药物关联信息和靶标关联信息, 集成生物网络与深度表示学习算法, 识别药物–靶标互作用。Zhang 等[25]利用自监督嵌入和异构聚合的多图卷积, 提取药物和靶标结构特征, 用于药物–靶标相互作用的预测。然而, 现有的智能算法无法充分提取异质生物网络中多尺度拓扑信息, 深度图神经网络在训练过程中容易产生梯度退化的相关问题。
针对药物–靶标互作用预测, 本文开发一种动态异质生物图表示的药物–靶标关系预测方法。该方法的框架主要由异质图数据表示、异质图门卷积和药物-靶标互作用预测器组成(图 1)。首先, 利用药物、蛋白质、疾病和副作用 4 种生物实体以及药物–药物对的邻接关系、药物–蛋白质对的邻接关系等 8 种边信息, 构建一个异质生物图; 然后, 为获得药物和靶标的低维嵌入信息, 在药物和靶标特征学习过程中需考虑不同类型节点特征的影响, 同时对多类型节点的邻域信息进行聚合。以 3 层异质图门卷积网络为例, 在第一层中, dr2 融合 dr1, dr3及节点在同质图中的特征, 同时融合异质图 pr2, pr5, di1 和se1 的节点特征; 在同质图中, pr3 融合 pr1, pr2 及节点特征; 在异质图中, 融合 dr1 和 di2 特征。最后, 拼接节点在每层学习到的特征, 利用内积(inner product, IP)函数预测 dr2 与 pr3 之间的关系评分。
本文构造的异质生物网络表示为 G=(V, E, TV, TE)。其中, V 表示异质生物网络中的节点集合, TV表示异质生物网络中的节点种类集合, 包括药物、蛋白质、疾病和副作用。TE 表示异质生物网络中边的种类集合, 包括药物–药物对的邻接关系、药物–蛋白质对的邻接关系、药物–疾病对的邻接关系、药物–副作用对的邻接关系、蛋白质–蛋白质对的邻接关系、蛋白质–疾病对的邻接关系、药物–药物对的相似度和蛋白质–蛋白质对的相似度。由于生物异质网络中节点和边的异质性, 不同种类的节点或边的属性特征会在不同的特征空间, 则节点特征矩阵表示为, 边的特征矩阵表示为。
基于药物–药物对的互作用、药物–副作用的关联信息和药物–疾病对的关联信息, 本文利用 Jac-card 相似系数计算药物之间的相似度网络。基于药物的化学结构, 利用 Tanimoto 系数[36]计算药物间的结构相似度。基于蛋白质疾病关联网络和蛋白质间互作用网络, 利用 Jaccard 相似系数计算蛋白质之间的相似性网络。利用双序列比对局部比对 Smith-Waterman 算法[37]计算蛋白质之间的序列相似度。在数据集的药物–药物对和蛋白质–蛋白质对的互作用网络, 药物–蛋白质对、蛋白质–疾病对和药物–副作用对的关联网络, 引入药物相似性信息和蛋白质相似性信息, 构建蛋白质、药物、疾病和副作用组成的异质生物图。与 Peng 等[7]构建二值网络方法类似, 药物、疾病和副作用关联矩阵生成过程的阈值分别为 0.5, 蛋白质关联矩阵生成过程的阈值为0.8。用蛋白质–疾病关联网络, 计算蛋白质–蛋白质相似性网络, 如果两个蛋白质之间的相似度(Si,j)大于 0.8, 则两个蛋白质之间关联值为 1, 反之为 0。蛋白质相似性二值的转换方式为
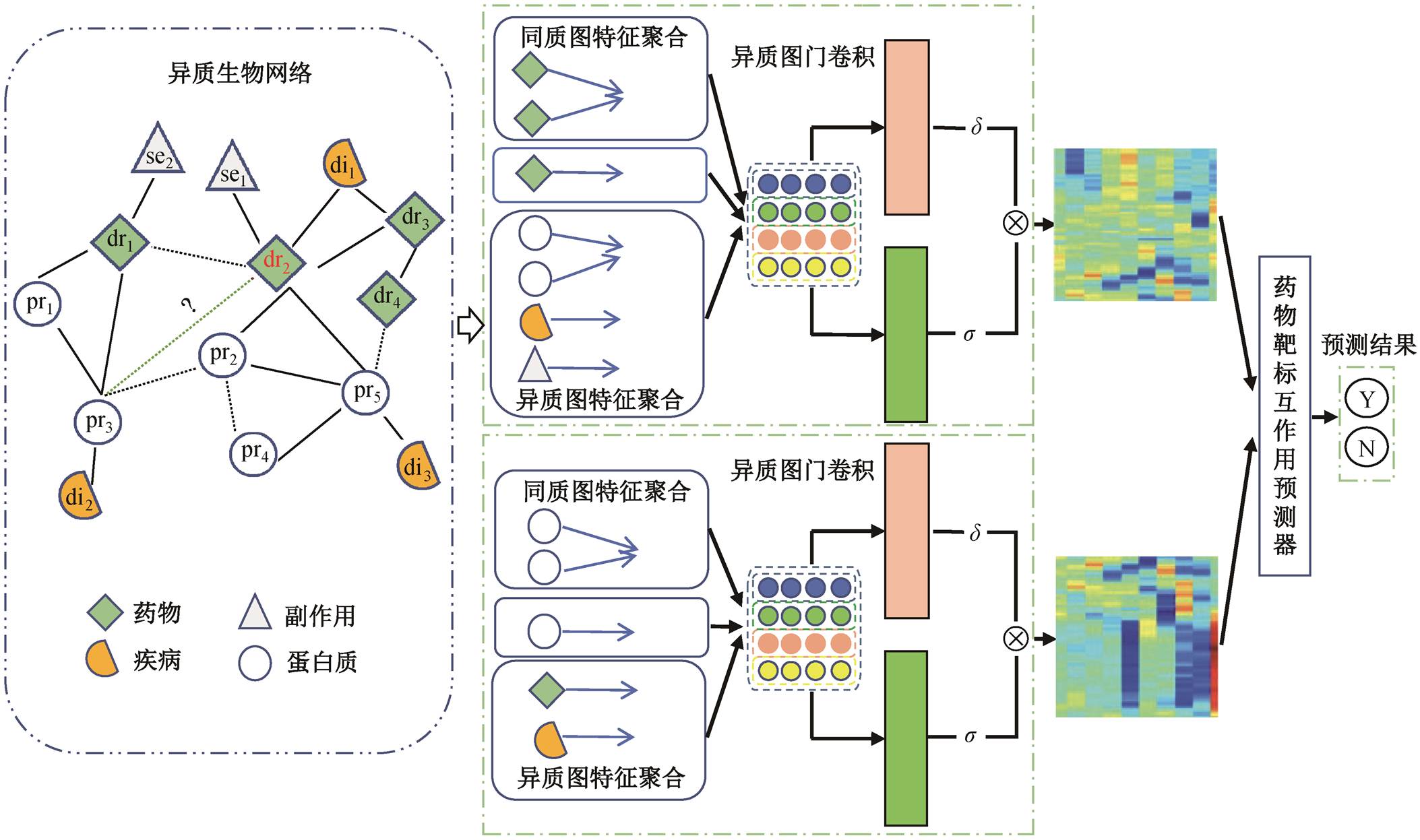
图1 动态异质生物图表示的药物靶标关系预测方法流程图
Fig. 1 Overall architecture diagram of drug target interaction prediction based on dynamic heterogeneous biological graph representation
 (1)
(1)药物、疾病和副作用等的二值相似性值转换规则为
 (2)
(2)
参照 Peng 等[7]的方法, 将构建的药物、疾病和副作用的二值相似性整合到药物互作用网络和蛋白质互作用网络中。若在药物–药物相互作用网络中, 药物 i与 j之间没有相互作用, 但其他药物–药物相似网络表明药物 i与 j高度相似, 则将药物 i和 j之间的相似边添加到药物–药物相互作用网络中。同样地, 对蛋白质的相似性网络进行类似处理, 并在蛋白质–蛋白质相互作用中添加一些额外的边。
本文设计的异质图门卷积是一种基于图卷积的指数型门控机制(图 1)。对于异质生物图中节点间的邻接关系类型 e,  是节点
是节点 的互作用邻接边类型为 e的邻居节点集合, 门控单元s( )和特征通道单元f( )分别表示异质图门卷积模型的门控机制和嵌入特征, 门控单元控制神经网络中特征通道单元的信息流动比例。对于药物靶标节点 V, 用节点和邻居节点特征向量进行聚合表示。具体地说, 在第一层, 模型药物靶标节点表示 V为初始向量 g0(V), 第 l层, 模型的药物靶标特征表示为 hl–1 (l≥2), 邻居节点的向量表示为
的互作用邻接边类型为 e的邻居节点集合, 门控单元s( )和特征通道单元f( )分别表示异质图门卷积模型的门控机制和嵌入特征, 门控单元控制神经网络中特征通道单元的信息流动比例。对于药物靶标节点 V, 用节点和邻居节点特征向量进行聚合表示。具体地说, 在第一层, 模型药物靶标节点表示 V为初始向量 g0(V), 第 l层, 模型的药物靶标特征表示为 hl–1 (l≥2), 邻居节点的向量表示为 。疾病和副作用节点
。疾病和副作用节点 由节点的初始向量表示
由节点的初始向量表示 构成。
构成。
为减少图神经计算的复杂性并增强模型的信息提取能力, 本文结合自动门控机制和图卷积, 设计一种高效的异质图门卷积模型 HGGCN, 用来提取药物和靶标的全局上下文依赖特征以及拓扑结构信息。通过 HGGCN 模型, 可以获得异构生物网络中药物、蛋白质、疾病和副作用节点在每一层的特征表示。药物或靶标的嵌入特征提取过程如下:
 (3)
(3)其中,  和
和 表示正则化系数;
表示正则化系数;  和
和 是第 l层图门卷积网络在边关系类型为 e时的学习参数; Ä是逐元素积融合运算, 用于融合门控单元s( )和特征通道单元f( )的信息;
是第 l层图门卷积网络在边关系类型为 e时的学习参数; Ä是逐元素积融合运算, 用于融合门控单元s( )和特征通道单元f( )的信息;  是节点 Vj 的邻接节点的特征,
是节点 Vj 的邻接节点的特征,  是节点Vi的特征, l=1 时
是节点Vi的特征, l=1 时
 ; s( )是 sigmoid 函数,
; s( )是 sigmoid 函数,  , 其神经元输出值范围是[0, 1];f( )是指数线性单元激活函数[38], 其神经元输出的计算定义为
, 其神经元输出值范围是[0, 1];f( )是指数线性单元激活函数[38], 其神经元输出的计算定义为
 (4)
(4)
其中, 参数a可控制神经元f输出值的饱和程度, 也可避免深度图神经网络训练中的梯度退化问题。在模型梯度反向传播更新的过程中, f( )的梯度计算定义为
 (5)
(5)本文模型 HGGCN 的梯度计算过程为
 (6)
(6)
式(6)表明该异质图门机制有一种独特的线性信息通路, 没有对门 部分进行缩放, 且可以加速梯度传播。该门单元具有非线性的特征转化能力, 在模型训练过程中, 可使深度异质图卷积模型在训练的梯度反向传播过程中减少梯度消散。
部分进行缩放, 且可以加速梯度传播。该门单元具有非线性的特征转化能力, 在模型训练过程中, 可使深度异质图卷积模型在训练的梯度反向传播过程中减少梯度消散。
对于异质生物图中的某个节点, 若异质图门卷积网络只有一层, 那么模型得到的节点特征表示只会聚合其邻居的一阶信息。因此, 多层图门卷积网络可以提取异质生物图中节点的高阶邻居特征、拓扑信息和语义特征。在模型 HGGCN 的每一层中, 每个节点的特征表示都通过聚合其邻居节点的特征来生成, 这些邻居节点由不同类型的边连接(图 1)。为了防止节点特征丢失并克服图卷积网络的过度平滑, 本文将不同层中每个节点的表示连接起来。具体地说, 给定一个药物或靶标在异构网络中, 对异质生物图中的药物或靶标节点 Vi经过多层异质图门卷积提取的特征, 其表示定义为
 (7)
(7)由于生物网络含有丰富的结构信息以及节点间的语义信息, 异质图中的药物和靶标等节点与药物疾病邻接等关联分别处于不同的特征空间中, 因此, HGGCN 将药物和靶标节点表示转换到低维向量空间中, 聚合异质图中不同邻接关系类型的节点信息, 提取多类型节点间的交互信息和语义信息, 得到包含异质图结构的药物和靶标节点特征嵌入。
通过我们设计的深度异质图门卷积网络, 从异质图中提取生物拓扑语义增强的药物特征表示为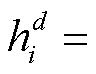
 靶标节点的特征为
靶标节点的特征为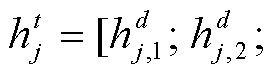
 。为了定量地估计药物 d(i)与靶标 t(j)间关联的概率, 将上述得到的药物和靶标的嵌入特征拼接, 通过对药物节点和靶标节点之间的一种内积(IP)运算, 得到的药物–靶标对之间存在关联的概率为
。为了定量地估计药物 d(i)与靶标 t(j)间关联的概率, 将上述得到的药物和靶标的嵌入特征拼接, 通过对药物节点和靶标节点之间的一种内积(IP)运算, 得到的药物–靶标对之间存在关联的概率为
 (8)
(8)鉴于动态异质生物图表示的药物–靶标关系预测方法的梯度计算和更新是可微的, 因此通过梯度下降方式训练和优化参数, 可以更好挖掘和推理药物靶标关系。当模型的目标函数是交叉熵损失函数时, 若输入数据为噪声, 也会得到非 0 即 1 的结果, 因此会增加过拟合的可能性。考虑到复杂的药物–靶标异质网络的特性, 因此模型的目标损失函数应增大类别间距。采用反向传播算法 Adam, 对整个模型进行端到端的训练, 优化模型参数并更新参数梯度。算法 1 展示 HGGCN 模型提取异质生物拓扑和语义信息的过程。
算法1 端到端的药物靶标互作用预测模型
输入药物、蛋白质、疾病、副作用。药物–药物对的互作用、药物–副作用对的关联网络和药物–疾病对的关联网络
输出优化后的模型及参数
S1 构建药物相似性网络
S2 构建靶标相似性网络
S3 引入药物相似性信息和蛋白质相似性信息构建蛋白质、药物、疾病和副作用组成的异质生物网络
S4 使用异质图门卷积网络计算节点和边之间的交互信息
S5 由式(3)更新每个药物、靶标节点中的特征、边特征
S6 构建药物靶标特征网络输出分子特征矩阵, 由式(7)~(8)得到预测结果
S7 由式(9)得到总损失函数。通过梯度下降优化更新模型参数
为更好地评估模型在不同参数下的损失, 并构建 HGGCN 模型来预测与实际数据的差异程度, 本研究设计一种基于交叉熵函数的铰链熵损失函数(hinge cross entropy loss, HCL), 用于评估 HGGCN模型进行药物–靶标互作用预测任务的损失值, 使得包含数据噪声的药物–靶标数据的异常预测比仅含正常推理的异常得分高, 目标函数计算过程可表示为
 (9)
(9)其中, yij表示药物 d(i)与靶标 t(j)间生物实验验证的关联标签。为了提高模型的泛化性能, 铰链熵损失函数可增大正样本预测概率, 减小负样本预测概率, 以此增大类间距, 使 HGGCN 模型在应对少量噪声输入时有一定的容错量。
本实验基于深度图库 Tensorflow 实现, 并采用Adam 作为模型 HGGCN 的优化器, 通过梯度下降法寻找一组最优的模型学习和推理参数。系统实验平台是 Ubuntu 20.04, 显卡是 NVIDIA GeForce RTX 3060(12G)。为了防止过拟合, 设置丢弃率(drop-out)为 0.2。本文模型的代码和数据可以通过 https: //gitlab.com/guoyb/hggcn 下载, 采用网格搜索的方式对超参数进行选择。为了验证本文方法的有效性和泛化能力, 在 DTINet 数据集[18]和 Yamanishi[36]数据集上评估药物–靶标互作用预测模型的性能, 然后进行对比分析和消融实验。DTINet 数据集记为数据集 1, 其中包含药物互作用网络、药物蛋白质互作用网络、药物疾病关联网络、药物副作用关联网络、蛋白质互作用网络和蛋白质疾病关联网络, 训练批次(Epochs)设置为 128, 学习率为 0.003, 隐藏层的维度大小为 64, 单次训练的样本个数 32, dropout 值为 0.2, 层数为 5。表 1 列出数据集 1 中 4种实体类型及数目、6 种边类型及每种类型边的数目。Yamanishi 数据集记为数据集 2, 包含酶(Enzy-me)、离子通道(ion channels, IC)、G 蛋白偶联受体(G-protein-coupled receptors, GPCR)和核受体(nuc-lear receptor, NR)4 种子数据集, 训练批次(Epochs)设置为 128, 学习率为 0.01, 隐藏层的维度大小为32, 单次训练的样本数为 32, dropout 值为 0.3, 层数为 3。表 2 列出数据集 2 中每种子数据集的药物、蛋白质和药物蛋白质关联网络的统计信息。
本文将药物靶标相互作用预测视为一种二分类问题, 采用 10 折交叉验证法对模型的药物靶标互作用预测能力进行评估。评价指标采用受试者工作特征曲线下面积 AUROC(area under the receiver opera-ting characteristic curve)和精确率–召回率曲线下面积 AUPRC(area under the precision-recall curve)来评估二分类预测模型预测性能。ROC 曲线是不同阈值下真阳性率(TPR)和假阳性率(FPR)的变化关系曲线。AUROC 和 AUPRC 涉及的 TPR, FPR, 精确率(Pre)和召回率(Rec)的计算方法如下:
表1 数据集 1 的统计信息
Table 1 Statistics of Dataset 1

实体实体数目边类型边数目 药物 708药物–蛋白质关联边1923 蛋白质1512药物–药物关联边10036 疾病5603药物–疾病关联边199214 副作用4192药物–副作用关联边80164 蛋白质–蛋白质关联边7363 蛋白质–疾病关联边1596745
表2 数据集 2 的统计信息
Table 2 Statistics of Dataset 2

数据类型酶离子通道蛋白偶联受体核受体 药物 445 210 223 54 蛋白质 664 204 95 26 药物–蛋白质关联边 2261476 638 90 药物–药物关联边893319262339184 蛋白质–蛋白质关联边69401792 297 44
 (10)
(10)其中, NTP 是真阳性, NTN 是真阴性, NFP是假阳性, NFN是假阴性。AUROC 适用于各类正负样本相对平衡的模型。当正负样本的数量不平衡时, AUPRC 比AUROC 更敏感。
3.3.1 自适应门控机制对模型的影响
为验证自适应门控机制的有效性, 本文分别利用不同门机制, 自适应地提取药物–靶标的特征表示, 在数据集 1 上的实验结果如图 2 所示。HGTU表示 HGGCN 中特征通道单元f( )是双曲正切函数, HGRELU 表示 HGGCN 中特征通道单元f( )是线性整流函数, HGLU 表示 HGGCN 中特征通道单元f( )是线性函数, HGELU 表示 HGGCN 中特征通道单元f( )是高斯误差函数。
从图 2 可以看出, 本文模型 HGGCN 在数据集 1上的药物–靶标互作用预测性能指标 AUROC 和AUPRC, 比 4 种基准模型平均提高 0.68%和 0.90%, 至少提高 0.37%和 0.55%。HGLU 中特征通道单元是一种简单的线性转换, 缺乏对数据的非线性建模能力; HGTU, HGRELU 和 HGELU 特征通道单元在训练过程中容易发生梯度消散问题, 使得模型无法充分提取异质生物图中的拓扑结构及语义信息。本文提出的门控单元和特征通道单元构成的图门机制, 可以使 HGGCN 自适应地提取更多药物–靶标异质网络中复杂依赖信息, 得到图结构拓扑信息强化的节点判别性特征。此外, 本文开发的图门控机制可以通过较少的非线性运算实现, 降低梯度消失问题, 使得充分建模动态异质生物图中的拓扑结构及语义信息, 提高模型的药物靶标互作用预测能力。
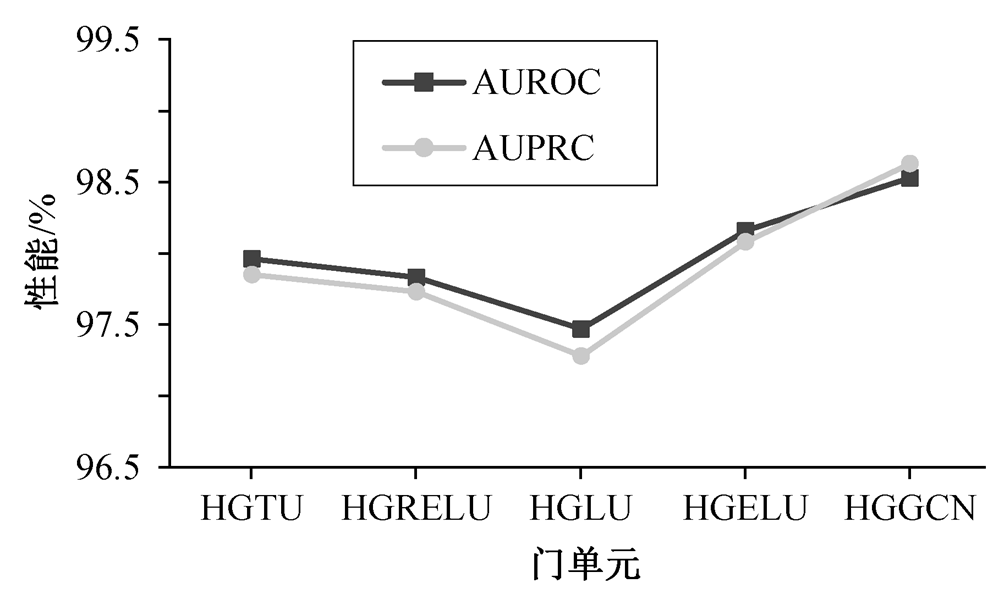
图2 不同门单元对 HGGCN 的性能比较
Fig. 2 Performance comparison of different gates for HGGCN
3.3.2 药物靶标互作用预测器对模型的影响
针对药物和靶标的复杂生物网络, 本文采用内积函数作为异质图门卷积网络 HGGCN 的预测器, 推理药物–靶标之间存在互作用的概率, 筛选药物–靶标预测任务的判别性模式信息。为验证内积(IP)函数预测器对药物–靶标互作用模型 HGGCN 推理能力, 分别利用 DistMult[39]、Bilinear[40]和 DEDI-COM 张量分解[15]预测器, 进行关联挖掘并推理预测药物与靶标互作用。在数据集 1 上的实验结果见图 3。
从图 3 可以看出, 与 3 种不同药物–靶标互作用判别器相比, 基于内积函数的异质图门卷积网络的性能指标 AUROC 和 AUPRC 平均分别提升 0.57%和0.55%。在内积函数的关联推理与预测下, 本文设计的异质图门卷积网络 HGGCN 可建模生物异质图中的多尺度长期依赖关系, 能提取异质图的层间和层内的图结构和语义特征, 强化并提高药物与靶标之间互作用预测模型的推理识别能力。
3.3.3 优化器对模型的影响
为了验证 Adam 优化器的有效性和优越性, 本文利用 SGD, Adamax, AdaGrad, RMSProp 和 NAdam等优化器调控 HGGCN 模型对药物和靶标的特征学习及关系推理预测能力, 在数据集 1 的实验结果如图 4 所示。可以看出, 与 5 种不同模型优化器相比, 利用 Adam 的异质图门卷积网络性能指标 AUROC和 AUPRC 平均分别提升 4.57%和 6.49%, 至少提高0.81%和 0.71%。Adam 优化器可以使 HGGCN 模型自动学习到最优参数, 进而提取到具有判别性信息的药物和靶标特征, 从而增强模型提取异质图的层间和层内的复杂图结构和丰富的语义特征以及药物–靶标预测的性能。
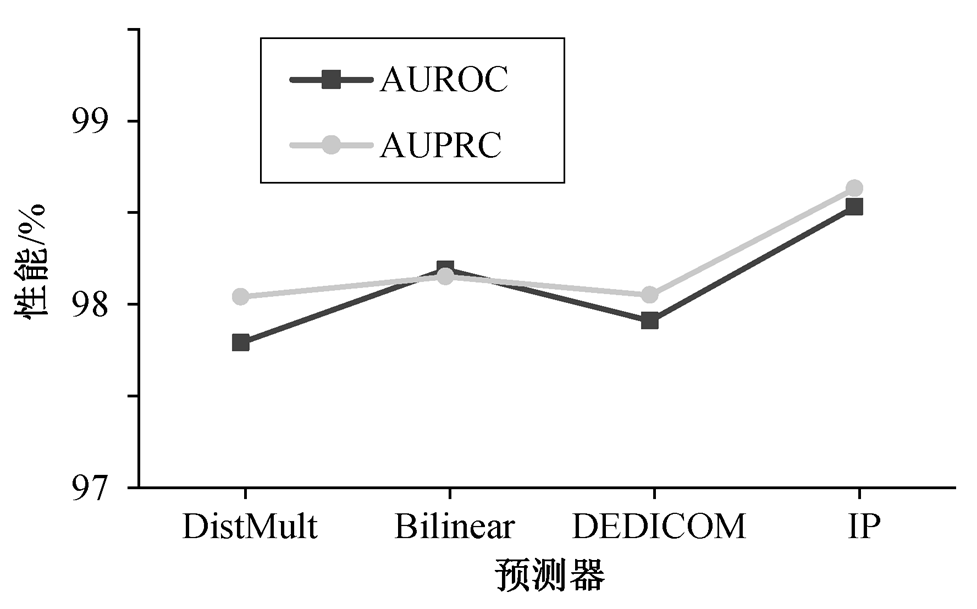
图3 不同预测器对 HGGCN 的性能比较
Fig. 3 Performance comparison of the different prediction functions for HGGCN
3.3.4 损失函数对模型的影响
本文将铰链熵损失函数 HCL 作为 HGGCN 模型预测药物–靶标互作用的目标函数。为对比损失函数对模型预测能力的影响, 分别利用合页损失函数(hinge loss, HL)以及交叉熵损失函数(cross entropy, CE)进行实验。HGGCN 在数据集 1 上的实验结果如表 3 所示。可以看出, 与两种不同损失函数优化的异质图门卷积网络相比, 利用铰链熵损失函数 HCL的指标 AUROC 和 AUPRC 平均分别提升 1.33%和1.64%, 至少提高 0.90%和 0.70%。结果表明, 损失函数 HCL 能够优化本文设计的动态异质生物图表示的药物–靶标关系预测方法, 使模型能够充分提取异质图中不同生物分子节点间的语义关联和长距离依赖特征, 有效地提升药物与靶标互作用挖掘的性能。
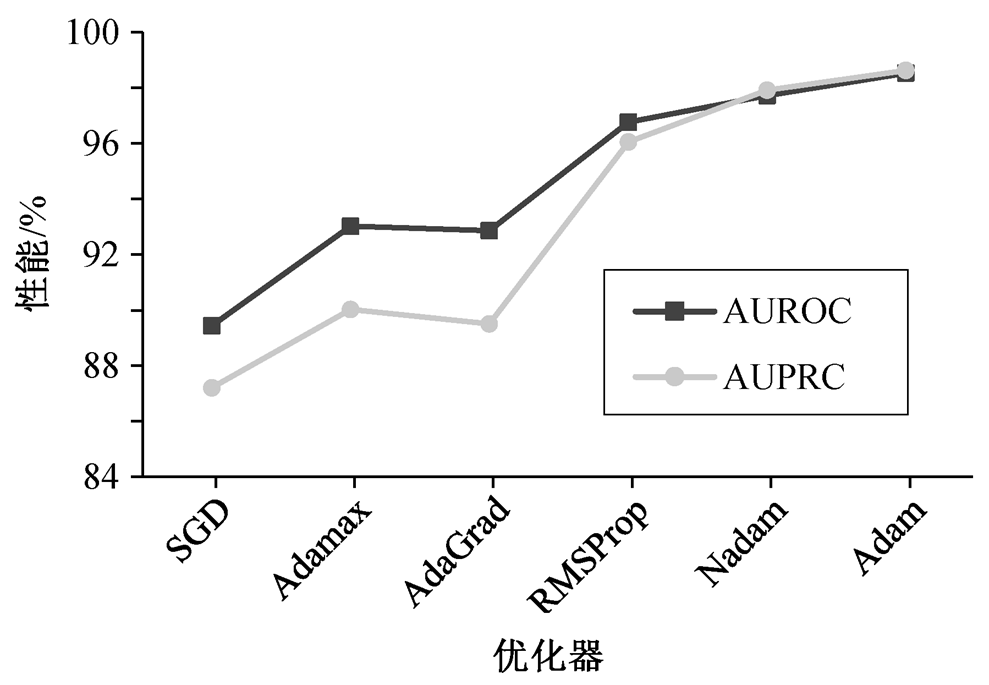
图4 不同优化器对 HGGCN 的性能比较
Fig. 4 Performance comparison of the different optimizers for HGGCN
表3 不同损失函数在数据集 1 上的消融实验结果
Table 3 Results of different loss functions for HGGCN on Dataset 1

模型损失函数AUROC/%AUPRC/% 基准模型HL96.7796.05 CE97.6397.93 本文模型HCL98.5398.63
3.4.1 基准模型
为了进一步评估模型预测的有效性和优越性, 本文将 HGGCN 模型在 Yamanishi 数据集和 DTINet数据集的预测结果与 13 种先进的药物–靶标关联预测方法进行比较。
1 )GAEN[22]是一种药物–靶标相互作用预测方法, 利用变分自编码器, 通过双瓦瑟斯坦生成对抗网络和梯度惩罚策略, 生成边缘信息和增强特征表示。
2 )DSGDTI[8]利用异构图自动编码器和基于异构注意网络的矩阵补全来预测药物靶标互作用。
3 )MIDTI[26]以无监督的方式构建整合药物和靶标的多视图相似性, 采用多视图相似网络融合和深度交互注意机制预测药物–靶标相互作用。
4 )GSRFDTI[23]整合多个药物关联信息和靶标关联信息, 集成生物网络与深度表示学习算法来识别药物–靶标互作用。
5 )MSHDTI[25]利用自监督嵌入和异构聚合的多图卷积提取药物和靶标结构特征, 用于药物–靶标相互作用的预测。
6 )BLMNII[24]是一种基于二部局部邻居交互轮廓的药物–靶标关系推断模型, 允许具有巨大相似性的邻居进行贡献, 引入高斯函数, 根据邻居与查询中药物与靶标的距离, 逐渐降低邻居节点的影响。
7 )NetLapRLS[35]是一种半监督学习方法——拉普拉斯正则化最小二乘法, 整合已知药物–蛋白相互作用网络信息以及化学结构和基因组序列数据, 预测药物–靶标关联信息。
8 )CMF[21]是一种多相似度协同矩阵因子分解模型, 将药物和靶标投影到一个共同的低秩特征空间中, 使其与药物和靶标的加权相似度矩阵相一致, 然后预测药物靶标关系。
9 )DTINet[18]是一个基于网络扩散的药物–靶标相互作用预测的机器学习算法, 通过构建一种异构网络, 学习药物–靶标关联信息并推断关联。
10 )EEGDTI[7]是一种新型的端到端图卷积表示学习框架, 利用多种实体和多种边缘类型的特征, 用于识别药物与靶标之间的相互作用。
11 )MGMADTI[20]是一种基于药物 SMILES 线的分子图和蛋白质氨基酸序列, 利用多阶门控卷积和多注意力机制融合开发的药物–靶标相互作用预测模型。
12 )MGNDTI[19]是一种基于多模态表示学习和门控机制的多模态预测模型, 利用不同的保留网络学习药物和靶标的序列表示。
13 )GraphDTA[29]是一种基于图卷积网络的药物靶标亲和力预测模型, 将药物表示为图, 并使用图神经网络来预测药物与靶标的亲和力。
3.4.2 性能对比分析
表 4 对比本文 HGGCN 模型和 13 种基准方法在数据集 1 上的药物靶标关系预测性能。可以看出, 本文 HGGCN 模型优于所有基准模型, AUPRC 和AUROC 指标平均分别提升 4.84%和 4.67%, 至少提高 0.35%和 0.24%。由于基准模型没有利用复杂生物网络的拓扑结构信息和语义信息, 缺少动态的特征选择和控制机制, 无法智能地筛选药物–靶标预测任务的判别性模式信息, 所以药物–靶标关系计算和预测效果均不理想。因此, 本文的药物–靶标关系预测方法采用图门卷积聚合异质邻居顶点特征, 将聚合后的特征与中心顶点的特征相融合, 可获得判别性的药物和靶标动态异质生物图表示, 可以很好地建模复杂生物网络、提取药物和靶标的嵌入特征, 推理并预测药物–靶标关系。
表4 在数据集 1 上模型的药物靶标关系预测性能对比
Table 4 Results of different models for drug-target interactions prediction on Dataset 1
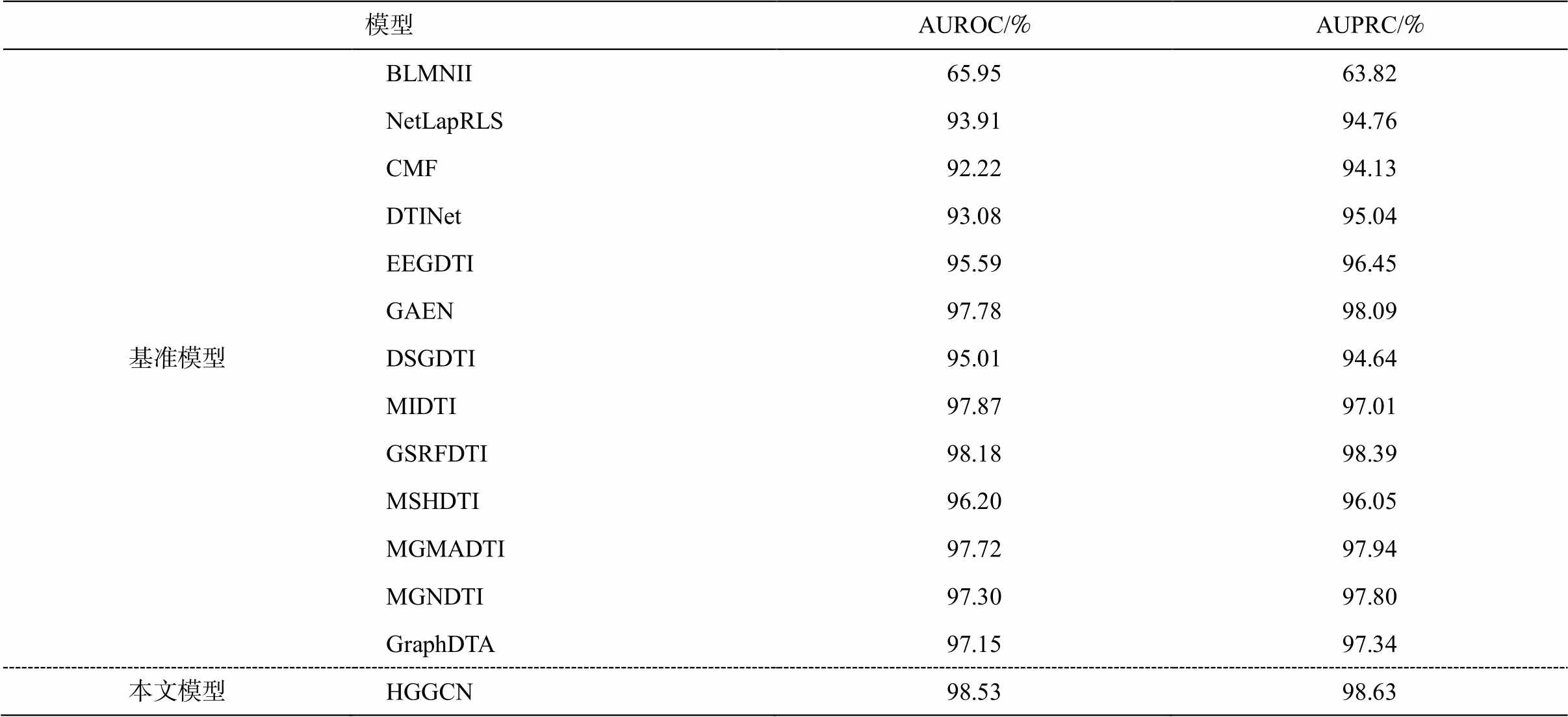
模型AUROC/%AUPRC/% 基准模型BLMNII65.9563.82 NetLapRLS93.9194.76 CMF92.2294.13 DTINet93.0895.04 EEGDTI95.5996.45 GAEN97.7898.09 DSGDTI95.0194.64 MIDTI97.8797.01 GSRFDTI98.1898.39 MSHDTI96.2096.05 MGMADTI97.7297.94 MGNDTI97.3097.80 GraphDTA97.1597.34 本文模型HGGCN98.5398.63
为验证 HGGCN 模型对生物异质图网络语义和拓扑信息建模能力和泛化能力, 选择现有 DTINet, EEGDTI, GSRFDTI, MIDTI, GraphDTA, MGMADTI和 MGNDTI 等先进的图表示学习算法, 分别在数据集 2 的 4 个子数据集(GPCR, Enzyme, NR 和 IC)上测试药物–靶标关系推理的性能。表 5 列出模型在数据集 2 上药物–靶标关系预测性能。可以看出, 在所有数据集上, HGGCN 模型的 AUPRC 和 AUROC 指标均优于所有基准模型。在 GPCR 数据集上, HGG-CN 模型的 AUPRC 和 AUROC 指标分别平均提升2.44%和 2.75%, 在 Enzyme 数据集上, 分别平均提升 2.49%和 1.89%, 在 NR 数据集上, 分别平均提升3.57%和 1.60%, 在 IC 数据集上, 分别平均提升2.41%和 2.21%。
表5 数据集 2 上模型的药物靶标关系预测性能对比
Table 5 Results of different models for drug-target interactions prediction on Dataset 2

模型数据集类型AUROC/%AUPRC/% 基准模型DTINetGPCR88.3387.89 Enzyme93.8094.79 NR82.8473.08 IC91.3990.89 EEGDTIGPCR96.0695.22 Enzyme98.8098.58 NR89.8889.36 IC98.6198.46 GSRFDTIGPCR96.4196.39 Enzyme97.5697.92 NR88.5083.92 IC98.0898.27 MIDTIGPCR96.8996.31 Enzyme97.8398.11 NR89.6884.92 IC98.3598.16 GraphDTAGPCR95.5996.62 Enzyme96.7097.38 NR86.0687.18 IC97.2597.66 MGMADTIGPCR96.5097.31 Enzyme97.9898.23 NR89.3689.76 IC97.7198.08 MGNDTIGPCR96.2596.06 Enzyme97.9098.19 NR90.2890.63 IC98.0998.29 本文模型HGGCNGPCR97.5997.86 Enzyme99.7199.49 NR91.6687.15 IC99.4899.33
对比实验结果表明, 本文提出的动态异质生物图表示的药物–靶标关系预测方法可以更好建模药物与靶标之间的复杂作用关系, 提取生物网络的拓扑和语义信息; 可以较好地建模不同类型节点之间的交互信息, 增强复杂网络中药物和靶标节点的判别性信息; 能够提供一种特征选择和控制机制, 智能筛选药物靶标预测任务的判别性模式。
针对药物靶标关系挖掘和推理任务, 本文提出一种拓扑结构及语义信息增强的动态异质生物图表示学习方法 HGGCN。首先, 将门控机制引入药物靶标异质网络的特征学习中, 构建一种图门控线性单元, 控制模型层间信息流和梯度传递, 缓解梯度随误差反向传播的退化问题; 然后, 集成药物和蛋白质特征以及经实验验证的药物互作用、药物蛋白质关联、药物疾病关联、药物副作用、蛋白质互作用和蛋白质疾病关联, 利用图门卷积聚合异质邻居顶点特征, 获得药物和靶标在异质图中药物–靶标的局部拓扑空间语义和长期序列的组合特征; 最后, 设计一种铰链熵损失函数, 进行模型参数学习和模型梯度更新。
本文在两个数据集上分别进行药物–靶标互作用挖掘实验。结果显示, 本文模型 HGGCN 有效地提升了药物与靶标的知识表示以及药物–靶标互作用挖掘任务的性能, 可作为药物–靶标间潜在关联预测的可靠工具。后续研究中, 我们将在异质图神经网络模型中嵌入更多多源信息, 如基因、生物通路、miRNA 序列信息和靶基因信息等, 并研究量子生物序列表示算法, 以期进一步提升模型的预测性能力和可解释性。
参考文献
[1] 鱼亮, 赵晋. 基于组织特异性和直接邻居相似度方法预测疾病–药物关系. 中国科学: 信息科学, 2019, 49(7): 1175–1185
[2] 刘健, 顾扬, 程玉虎, 等. 基于多智能体强化学习的乳腺癌致病基因预测. 自动化学报, 2022, 48(6): 1246–1258
[3] 饶晓洁, 张通, 孟献兵, 等. 基于多层注意力和消息传递网络的药物相互作用预测方法. 自动化学报, 2022, 48(1): 1–13
[4] 丁苍峰, 王君, 张紫芸. 多层异构生物网络候选疾病基因识别. 自动化学报, 2022, 48(2): 1–16
[5] 郭茂祖, 王诗鸣, 刘晓燕, 等. miRNA与疾病关联关系预测算法. 软件学报, 2017, 28(10): 3094–3102
[6] 廖懿鸣, 欧阳纯萍, 刘永彬, 等. 基于异质信息网络元路径的药物‒靶标相互作用预测模型. 北京大学学报(自然科学版), 2022, 58(1): 37–44
[7] Peng J, Wang Y, Guan J, et al. An end-to-end hetero-geneous graph representation learning-based frame-work for drug-target interaction prediction. Briefings in Bioinformatics, 2021, 22(3): 1–12
[8] Li Y, Liang W, Peng L, et al. Predicting drug-target interactions via dual-stream graph neural network. IEEE/ACM Transactions on Computational Biology and Bioinformatics, 2024, 21(3): 948–958
[9] Chu Z, Huang F, Fu H, et al. Hierarchical graph representation learning for the prediction of drug- target binding affinity. Information Sciences, 2022, 613: 507–523
[10] Ye X B, Guan Q, Luo W, et al. Molecular substructure graph attention network for molecular property identi-fication in drug discovery. Pattern Recognition, 2022, 128: 108659
[11] Lee H, Lee J W. Target identification for biologically active small molecules using chemical biology app-roaches. Archives of Pharmacal Research, 2016, 39 (9): 1193–1201
[12] 吴越, 王英, 王鑫, 等. 基于超图卷积的异质网络半监督节点分类. 计算机学报, 2021, 44(11): 2248–2260
[13] 高创, 李建华, 季秀怡, 等. 基于图卷积神经网络的药物靶标作用关系预测方法. 计算机科学, 2021, 48(6): 127–134
[14] Xu H, Sang S, Bai P, et al. GripNet: Graph information propagation on supergraph for heterogeneous graphs. Pattern Recognition, 2023, 133: 108973
[15] Yang X, Wang W, Ma J L, et al. BioNet: a large-scale and heterogeneous biological network model for inter-action prediction with graph convolution. Briefings in Bioinformatics, 2022, 23(4): bbab491
[16] Zitnik M, Agrawal M, Leskovec J. Modeling polyphar-macy side effects with graph convolutional networks. Bioinformatics, 2018, 34(13): i457–i466
[17] Mei J, Kwoh C K, Yang P, et al. Globalized bipartite local model for drug-target interaction prediction // Proceedings of the 11th International Workshop on Data Mining in Bioinformatics. Beijing, 2012: 8–14
[18] Luo Y, Zhao X, Zhou J, et al. A network integration approach for drug-target interaction prediction and computational drug repositioning from heterogeneous information. Nature Communications, 2017, 8: no. 573
[19] Peng L, Liu X, Chen M, et al. MGNDTI: a drug-target interaction prediction framework based on multimodal representation learning and the gating mechanism. Journal of Chemical Information and Modeling, 2024, 64(12): 6684–6698
[20] Li C, Mi J, Wang H, et al. MGMA-DTI: drug target interaction prediction using multi-order gated convolu-tion and multi-attention fusion. Computational Biology and Chemistry, 2025, 118: 108449
[21] Zheng X, Ding H, Mamitsuka H, et al. Collabora- tive matrix factorization with multiple similarities for predicting drug-target interactions // Proceedings of the 19th ACM SIGKDD International Conference on Knowledge Discovery and Data Mining. Chicago, 2013: 1025–1033
[22] Li G, Sun W, Xu J, et al. GA-ENs: a novel drug–target interactions prediction method by incorporating prior knowledge graph into dual Wasserstein generative ad-versarial network with gradient penalty. Applied Soft Computing, 2023, 139: 110151
[23] Zhu Y, Ning C, Zhang N, et al. GSRF-DTI: a frame-work for drug-target interaction prediction based on a drug-target pair network and representation learning on a large graph. BMC Biology, 2024, 22: 156
[24] Mei J P, Kwoh C K, Yang P, et al. Drug-target interac-tion prediction by learning from local information and neighbors. Bioinformatics, 2013, 29(2): 238–245
[25] Zhang B, Niu D, Zhang L, et al. MSH-DTI: multi-graph convolution with self-supervised embedding and he-terogeneous aggregation for drug-target interaction prediction. BMC Bioinformatics, 2024, 25: 275
[26] Song W, Xu L, Han C, et al. Drug-target interaction predictions with multi-view similarity network fusion strategy and deep interactive attention mechanism. Bioinformatics, 2024, 40(3): btae346
[27] Tian Q, Bilgic B, Fan Q, et al. DeepDTi: high-fidelity six-direction diffusion tensor imaging using deep lear-ning. NeuroImage, 2020, 219: 117017
[28] 孙明龙, 欧阳纯萍, 刘永彬, 等. 基于分层融合策略和上下文信息嵌入的多模态情绪识别. 北京大学学报(自然科学版), 2024, 60(3): 393–402
[29] Nguyen T, Le H, Quinn T P, et al. GraphDTA: pre-dicting drug-target binding affinity with graph neural networks. Bioinformatics, 2020, 37(8): 1140–1147
[30] Guo Y, Pang D, Huang M, et al. Deep dual dynamic gated convolutional model with interpretable pooling for genomic signal prediction [J/OL]. Data Intelligen-ce, (2025–03–01) [2025–03–04]. https://doi.org/10.3724/ 2096-7004.di.2025.0051
[31] Lounkine E, Keiser M J, Whitebread S, et al. Large-scale prediction and testing of drug activity on side-effect targets. Nature, 2012, 486: 361–367
[32] Öztürk H, Ozkirimli E, Özgür A. A novel methodology on distributed representations of proteins using their interacting ligands. Bioinformatics, 2018, 34(13): i295– i303
[33] Thomsen R, Christensen M H. MolDock: a new techni-que for high-accuracy molecular docking. Journal of Medicinal Chemistry, 2006, 49(11): 3315–3321
[34] Goodsell D S, Morris G M, Olson A J. Automated docking of flexible ligands: applications of AutoDock. Journal of Molecular Recognition, 1996, 9(1): 1–5
[35] Xia Z, Wu L Y, Zhou X, et al. Semi-supervised drug-protein interaction prediction from heterogeneous bio-logical spaces. BMC Systems Biology, 2010, 4 (Suppl 2): S6
[36] Yamanishi Y, Araki M, Gutteridge A, et al. Prediction of drug–target interaction networks from the integra-tion of chemical and genomic spaces. Bioinformatics 2008; 24(13): i232–i240
[37] Abu-Hashem M, Gutub A. Efficient computation of Hash Hirschberg protein alignment utilizing hyper th-reading multi-core sharing technology. CAAI Transac-tions on Intelligence Technology, 2022, 7(3): 278–291
[38] Clevert D A. Fast and accurate deep network learning by exponential linear units (ELUs) [EB/OL]. (2016–02–22)[2025–09–29]. https://doi.org/10.48550/arXiv. 1511.07289
[39] Schlichtkrull M, Kipf T N, Bloem P, et al. Modeling relational data with graph convolutional networks // European Semantic Web Conference. Cham: Springer, 2018: 593–607
[40] Fan Y, Chen M, Pan X, et al. GCRFLDA: scoring lncRNA-disease associations using graph convolution matrix completion with conditional random field. Brie-fings in Bioinformatics, 2022, 23(1): bbab361
Drug-target Interaction Prediction Based on Dynamic Representation Learning of Heterogeneous Biological Graphs
Abstract To extract the complex relationships between drugs and targets, this paper designs a dynamic representation learning algorithm based on deep heterogeneous graph gated convolutional networks (HGGCN) for biological graph modeling and representation learning. The algorithm combines the merits of the gated channels and feature channels to adaptively model interaction patterns of heterogeneous graphs, enhance the topological structure and semantic information of complex networks based on the fusion, and obtain the discriminative representation of drugs and targets for drug-target interaction mining. Experimental results show that the proposed model outperforms existing drug target interaction prediction methods, and is also an accurate drug target association prediction tool, which could provide the technical support for the precision treatment of complex diseases and network information mining.
Key words complex biological networks; multi-source heterogeneous graph; neural networks; drug-target inter-action