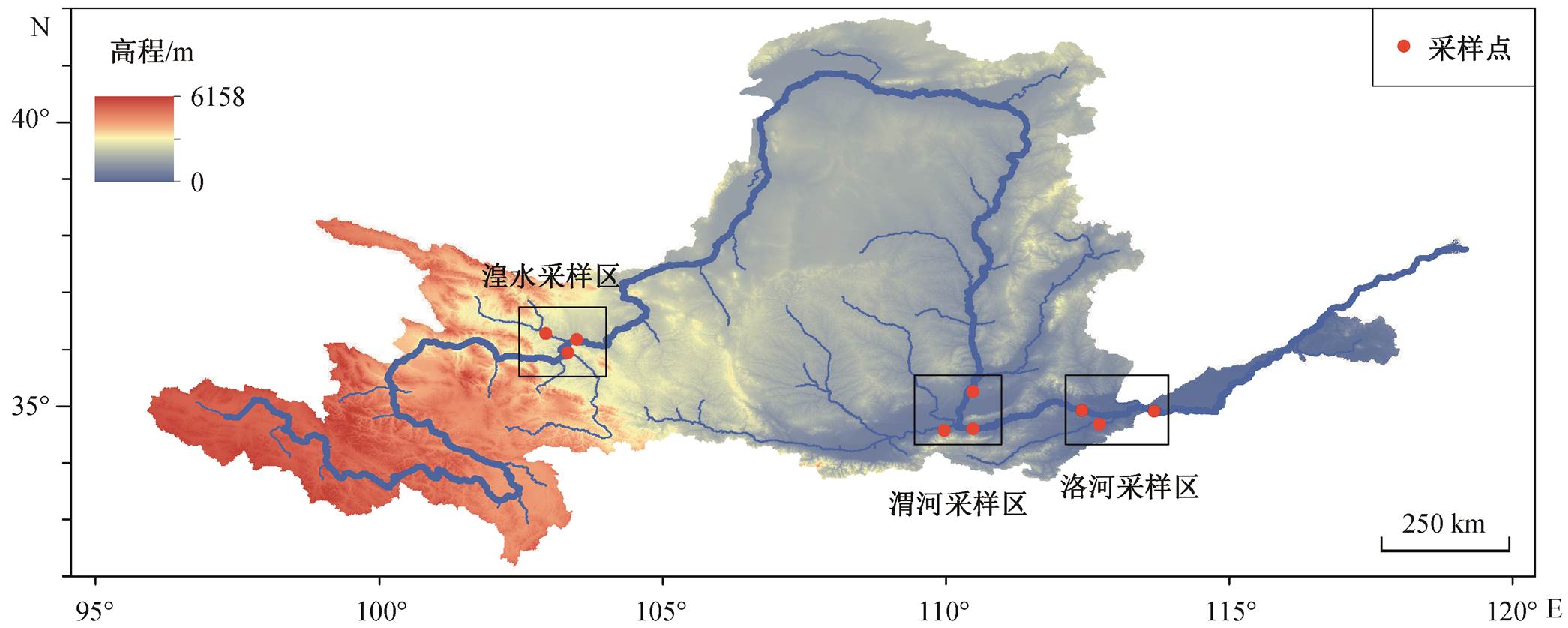
图1 研究区域采样点
Fig. 1 Sampling sites in the study area
北京大学学报(自然科学版) 第60卷 第2期 2024年3月
Acta Scientiarum Naturalium Universitatis Pekinensis, Vol. 60, No. 2 (Mar. 2024)
doi: 10.13209/j.0479-8023.2023.060
国家自然科学基金(52100171)和深圳市科技研发资金(20220808011114001)资助
收稿日期: 2023–02–16;
修回日期: 2023–03–18
摘要 利用宏基因组学技术, 分析黄河典型入干支流湟水河、渭河和洛河水体及沉积物中噬菌体的群落结构, 并对三条支流的噬菌体多样性、宿主和功能潜能进行比较。结果显示, 从宏基因组中识别出的 99.77%的未知 vOTUs 无法与 NCBI Refseq 参考数据库聚类, 表明噬菌体群落的物种新颖度较高。检出的噬菌体隶属 15 科 158 属, 湟水河中特有的噬菌体属最多, Pbi1virus, Badnavirus, Ea214virus 和 Gammabaculovirus 仅在该支流中检出。3 条支流的噬菌体群落结构具有显著差异, 且沉积物中群落的空间异质性更大。多种辅助代谢基因(AMGs)表明噬菌体可能参与了湟水河、渭河及洛河采样区域的氮、碳和磷等元素的循环过程, 但 3 条支流中的 AMGs 分布无差异。噬菌体群落的宿主范围跨越 18 个原核生物门, 以变形菌门、放线菌门和拟杆菌门为主, 1.8%的噬菌体具有跨门感染的潜在能力。洛河的水体及沉积物中富集了感染 Planctomycetota 的噬菌体, 渭河沉积物样本中感染Chloroflexota 的噬菌体含量较高。细菌群落和噬菌体群落存在显著的相关性, 且水体中噬菌体与其宿主的一致性比沉积物更高。
关键词 支流; 病毒宏基因组学; 噬菌体群落; 群落结构特征
作为一种专性寄生细胞生命的生物实体, 病毒在地球上有着最丰富的多样性, 且广泛存在于各种生态系统中[1]。病毒能够感染并裂解宿主, 从而调控细胞宿主的丰度和群落结构[2]。宏基因组测序技术突破了传统研究中微生物难以培养的局限, 逐渐成为病毒学深入研究的重要手段[3]。环境病毒宏基因组学已经广泛应用于探索海洋[4–5]、土壤[6–7]、湖泊[8]、极地[9]以及人类肠道[10]等生态系统。未培养的病毒序列(uncultivated virus genomes, UViGs)对公开数据库的贡献已超过 95%[11], 目前最大的 UViGs数据库 IMG/VR v4 包含超过 1500 万个的病毒基因组和 870 万个 vOTUs[12], 极大地拓展了人们对环境病毒的分布及其遗传多样性的认识。基于宏基因组学方法和计算机预测手段, 可以为病毒预测到更广泛的宿主范围[13], 并揭示病毒与其宿主间的动态关系[14]。对海洋[15]和土壤[7]等生态系统的研究表明, 病毒编码了各种核心结构基因以及辅助代谢基因(AMGs), 能够参与宿主细菌的中心碳代谢、磷酸盐代谢和硫循环等过程[16–17]。
河流是地球生态的重要参与者, 对大陆和海洋生态产生深远而复杂的影响, 探究河流微生物群落的多样性能够为维护河流绿色生态功能提供重要参考。黄河是我国第二长河, 先后有 13 条主要支流汇入, 向干流补充水量, 输送泥沙[18]。支流对干流的净流量[19]、水温[20]和水质[21]等环境因子产生影响。研究人员已经对黄河流域的浮游藻类[22]、细 菌[23]等微生物群落及抗性基因[24]展开调查, 并探究干支流的微生物群落差异。对黄河主要支流渭河及其支流的研究结果表明, 渭河中浮游细菌群落的系统发育多样性指数显著高于其支流[25]。对汾河入黄口的研究结果表明, 汾河与黄河的微生物差异性大于汾河内以及黄河内各样点之间的微生物差异性, 汾河水的汇入导致交汇区下游黄河微生物结构差异明显[26]。但是, 目前对黄河典型支流水体及沉积物中噬菌体的研究相对缺乏, 对其群落结构与特征尚不了解。
基于上述背景, 本研究于 2015 年 5 月在渭河、洛河和湟水 3 条黄河典型支流的入干区采集水体和沉积物样本, 探究噬菌体群落结构、噬菌体编码的功能基因多样性以及噬菌体与细菌宿主群落的关系, 并对 3 条支流进行比较, 以期为河流微生物生态学研究提供参考。
渭河是黄河最大的支流, 于陕西省境内汇入干流; 洛河是黄河下游重要支流, 于河南省境内汇入干流; 湟水是黄河上游重要支流, 于甘肃省境内汇入干流。根据国家水文站点分布, 本研究在湟水(HS)、渭河(WH)和洛河(LH)这 3 条支流的入干区设置采样区, 每个采样区在支流上靠近汇入点处设置一个采样点, 在干流的汇入点前后两处各设置一个采样点(图 1), 于2015 年 5 月采集 9 个断面的水体(WSF)和沉积物(SS)样本共 17 个, 其中水体样本 9个, 沉积物样本 8 个。
采集的水样首先用 3.0μm孔径的聚碳酸脂滤膜(Millipore, USA)过滤, 所得滤液通过 0.22μm 孔径聚碳酸脂滤膜(Millipore, USA), 截留水体中直径为0.22~3.0μm 的颗粒作为水体样本。在每个站点水深约 0.5m 处同步采集表层沉积物, 储存于 50mL 灭菌离心管中, 作为沉积物样本。使用 FastDNA® SPIN Kit for Soil (MP Biomedicals, 美国)试剂盒, 参考试剂盒说明书进行 DNA 提取。每一个环境样本进行 3~5 次 DNA 抽提, 之后将每个环境样本的 3~5个 DNA 样品进行混合, 组成该环境样本的微生物DNA 样品。吸取 5μL 环境 DNA 样品, 用 0.8%的琼脂糖凝胶电泳检测 DNA 完整性。环境 DNA 样品用Nanodrop 2000 进行定量, 使用 Illumina Hiseq 进行宏基因组测序。
使用 metaWRAP[27]对原始 reads 进行质量控制, 并用 megahit 拼接序列。采用 PPR-Meta 等软件识别噬菌体, 使用 CheckV[28]预测噬菌体序列质量, 并用 CD-HIT[29]进行聚类。噬菌体序列的分类学注释使用 VPF-Class[30]完成。基于 vConTACT2, 将噬菌体序列进行共享蛋白网络分析, 并将结果可视化。使用 Prodigal[31]预测噬菌体序列的开放阅读框(open reading frames, ORF), 并用 eggNOG-mapper[32] 软件注释蛋白, 根据 COG 功能, 对结果进行进一步分类。对于原核生物群落, 使用 metaWRAP[27]的binning 模块对 contig 进行分箱, 生成宏基因组组装基因组(metagenomic assembly genomes, MAGs), 利用 GTDB-Tk[33]对每个 MAGs 进行分类学注释。
图1 研究区域采样点
Fig. 1 Sampling sites in the study area
使用 bowtie2[34]和 samtools[35]计算噬菌体和原核生物的丰度, 得到 RPKM 丰度值(每千碱基 reads每百万映射 reads)。基于序列相似性、CRISPR 间隔子序列匹配和 tRNA 结合位点匹配的原理[36–37], 从原核生物中预测噬菌体的假定宿主。
使用 shell 和 python 脚本完成宏基因组数据提取和格式转换等数据处理工作。计算噬菌体群落的丰富度、香农指数和辛普森指数等 alpha 多样性指数, 并计算样本间 Bray-Curtis 距离, 得到距离矩阵, 然后通过非度量多维尺度分析(nonmetric multidi-mensional scaling, NMDS)探究其群落分布特性, 采用相似性分析(analysis of similarity, ANOSIM)方法检验组间差异是否显著。使用 STAMP 软件, 基于Welch’s t 检验方法分析基因的分布差异性。使用Procrustes 分析得到噬菌体与宿主群落间相关性。上述分析及结果的可视化采用 R 语言(v3.6)的 vegan, picante 和 ggplot2 包完成。
经过聚类, 从 3 条支流中总共检出 111063 个vOTUs。同时, 根据整合酶等指示基因, 使用CheckV 对噬菌体基因组的感染模式进行预测, 仅41 个 vOTUs 被预测为溶原噬菌体, 占 0.3‰, 3 条支流中的噬菌体以高度活跃的裂解感染模式生存。
对 vOTUs 进行物种注释, 其中 37.0%的病毒得到 Baltimore 分类注释, 绝大部分 vOTUs 隶属双链DNA 噬菌体, 仅有 16 条序列被注释为单链 DNA 噬菌体, 占比低于 0.39‰。
科水平的噬菌体注释率为 32.3%, 共检出 15 个科(表 1), vOTUs 数量最多的科均隶属有尾噬菌体目, 其中, 长尾噬菌体科(Siphoviridae)有 11567 个, 短尾噬菌体科(Podoviridae)有 10791 个, 肌尾噬菌体科(Myoviridae)有 10716 个, 丰富度占比超过92.3%。本研究还鉴定出 2440 个 vOTUs 隶属藻类DNA 病毒科(Phycodnaviridae), 能够感染真核藻类, 在所有样本中相对丰度为 4.1%。河流为真核藻类生长提供环境, 黄河流域中已经检测出硅藻[38]等真核藻类, 为其提供大量宿主[39]。湟水河共检出 15科(沉积物 12 科, 水体 14 科), 渭河检出 13 科(沉积物 12 科, 水体 13 科), 洛河检出 14 科(沉积物 11 科, 水体 14 科)。
表1 vOTUs科水平注释
Table 1 Annotation of vOTUs at family level
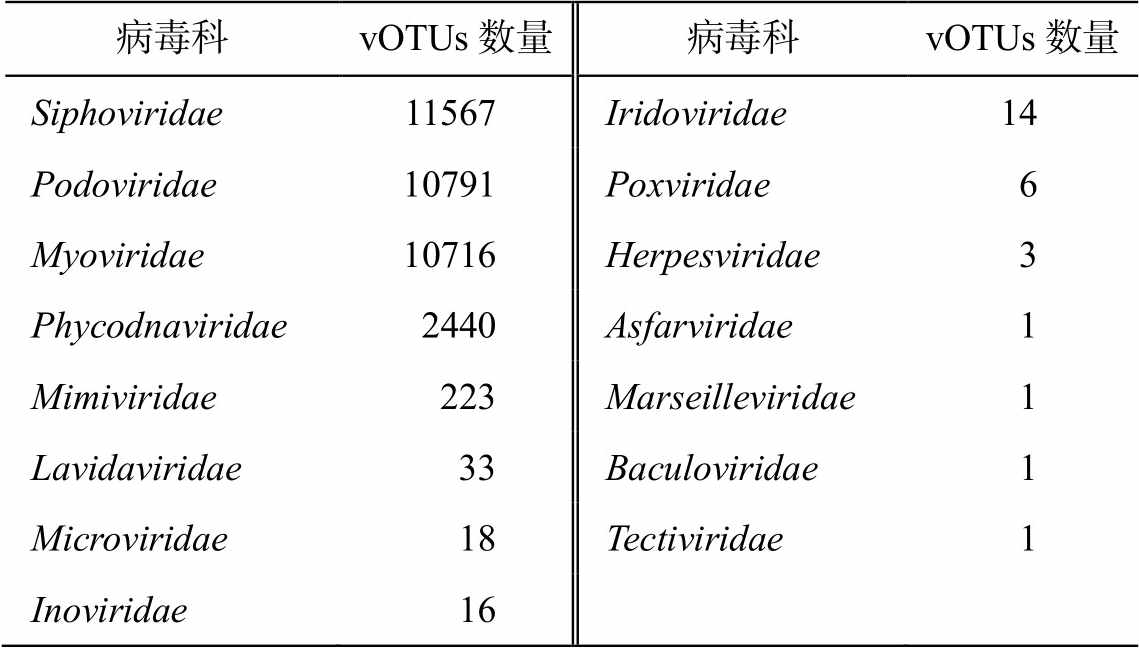
病毒科vOTUs数量病毒科vOTUs数量 Siphoviridae11567Iridoviridae14 Podoviridae10791Poxviridae6 Myoviridae10716Herpesviridae3 Phycodnaviridae2440Asfarviridae1 Mimiviridae223Marseilleviridae1 Lavidaviridae33Baculoviridae1 Microviridae18Tectiviridae1 Inoviridae16
属水平噬菌体注释率为 14.8%, 3 条支流中噬菌体隶属 158 个属, 数量最多的 5 个噬菌体属分别为Prymnesiovirus(2214 个), Lambdavirus(2040 个), T4virus(1714 个), P22virus(1483 个), P12024virus(1177 个)。湟水河检出 147 属(沉积物 114 属, 水体145 属), 渭河检出 153 属(沉积物 134 属, 水体 153属), 洛河检出 150 属(沉积物 135 属, 水体 149 属)。湟水河中特有的噬菌体属最多, Pbi1virus, Badna-virus, Ea214virus 和 Gammabaculovirus 仅在该支流样品中检出; Bacilladnavirus 仅在洛河沉积物中检出; Luz7virus仅在渭河水体中发现。
将 vOTUs 与 NCBI Refseq 数据库中的高质量参考病毒基因组进行比对, 基于共享基因网络, 最终形成 15451 个属水平的病毒簇(图 2), 其中 253 个vOTUs 与 NCBI Refseq 参考病毒基因组聚类, 仅占所有 vOTUs 的 0.23%。绝大多数 vOTUs 与参考病毒基因组的差异较大, 这些未知的噬菌体表明黄河典型支流入干区噬菌体群落具有较高的新颖性。
3 条支流中, 噬菌体科水平的分布具有明显差异(图 3(a))。相对而言, 沉积物中噬菌体组成的河流差异更大。Siphoviridae 在湟水河和渭河中含量较高(>40%), 显著高于洛河(26%); Podoviridae 在渭河和洛河中富集, 相对丰度超过 30%, 而在湟水河沉积物中仅为 16%; Myoviridae 在湟水河和洛河中含量较高, 相对丰度超过 30%, 而在渭河中仅占21%。水体样本中, Siphoviridae 在 3 条支流中差异不大, 相对丰度均超过 30%; Podoviridae 在渭河中富集(含量>36%), 在湟水河中含量最低(<25%); Myoviridae 在湟水河和洛河中富集, 相对丰度超过30%, 而在渭河中仅为 23%。
为探究 3 条支流噬菌体群落结构的差异, 基于Bray-Curtis 距离对 vOTUs 进行 NMDS 分析。如图4(a)所示, 水体和沉积物样本中的噬菌体群落有明显分离, 两种介质中噬菌体群落差异显著(P=0.02, R=0.489)。如图 4(b)和(c)所示, 沉积物和水体中, 3条支流的噬菌体群落结构差异显著, 表明地理空间对噬菌体群落也存在影响。沉积物群落(R=0.701)的支流间差异大于水体群落(R=0.432), 空间异质性作用更强。河流是单向流动的连续性系统[40], 水体微生物可随水流从上游输送至下游, 而沉积物在积年沉降作用下较为稳定, 从而产生更为显著的组间差异。
综上所述, 湟水河、渭河和洛河 3 条支流在噬菌体群落组成和结构上具有显著差异。湟水河主要流经青海和甘肃, 多年平均径流量为 16.2 亿 m3, 流域内气温较低, 属于干旱半干旱大陆性气候[41]; 渭河主要流经甘肃和陕西, 年径流量为 100.5 亿 m3, 年输沙量超过 5 亿吨, 是向黄河输送水流、泥沙最多的支流[42]; 洛河主要流经陕西和河南, 年均流量为 34.3 亿 m3, 属于温带季风性气候。湟水河、渭河、洛河在气候地理和水文环境方面具有较大的差别, 对微生物群落的塑造产生影响, 从而导致噬菌体群落结构的差异。此外, 3 条支流中检测出大量环境噬菌体物种, 它们随着水流和泥沙的输移进入干流, 是干流中噬菌体物种多样性的重要贡献者。
对预测出的噬菌体 ORF 进行 COG 功能注释, 检测到 267411 个 ORF, 从属于 23 个 COG 功能类别, 展示了高度的功能多样性(图 5(a))。除 S 类功能未知的蛋白外, 平均每个样本中有超过 4000 个 ORF编码如下类别的蛋白: L 复制/重组和修复、M 细胞壁/膜/包膜的生物合成、K 转录、O 转录后修饰, 证实噬菌体群落编码的蛋白质主要用于维持繁殖和转录等过程。
噬菌体可以通过 AMGs 的掺入和表达来影响微生物种群的多样性和功能[6], 从广义上讲, AMGs 指噬菌体中所有涉及代谢的基因, 包括能量的产生和转化类(C 类)、氨基酸的运输和代谢类(E 类)、核酸的运输和代谢类(F 类)、碳水化合物的运输和代谢类(G 类)、辅酶的运输和代谢类(H 类)、脂质的运输与代谢类(I 类)以及无机离子的运输和代谢类(P类)[17]。本研究共检测出 20213 个 AMGs, 占功能基因总数的 7.6%, 其中 C 类有 1278 个, E 类有 3687 个, H 类有 4611 个, F 类有 6674 个, G 类有 5899 个, I 类有 1307 个, P 类有 1368 个, 平均每个样本中有 1189个 ORF 编码 AMGs, 涉及核酸、碳水化合物和氨基酸代谢过程的基因最多, 展现较大的辅助代谢潜力, 对湟水河、渭河及洛河区域的氮、碳和磷等元素循环过程具有潜在影响。
如图 5(b)所示, 部分 AMGs 在沉积物和水体中的分布具有差异。沉积物中富集 P 类基因, 更多的噬菌体参与同化硫酸盐还原、铁储存等无机盐运输和代谢过程; 水体样本中富集 G 类基因, 更多的噬菌体参与纤维素水解、核苷酸焦磷酸水解等碳水化合物代谢过程。对 3 条支流的基因丰度进行比较, 未发现 AMGs 的分布具有地域差异, 湟水河、渭河及洛河的辅助代谢潜能较为接近。
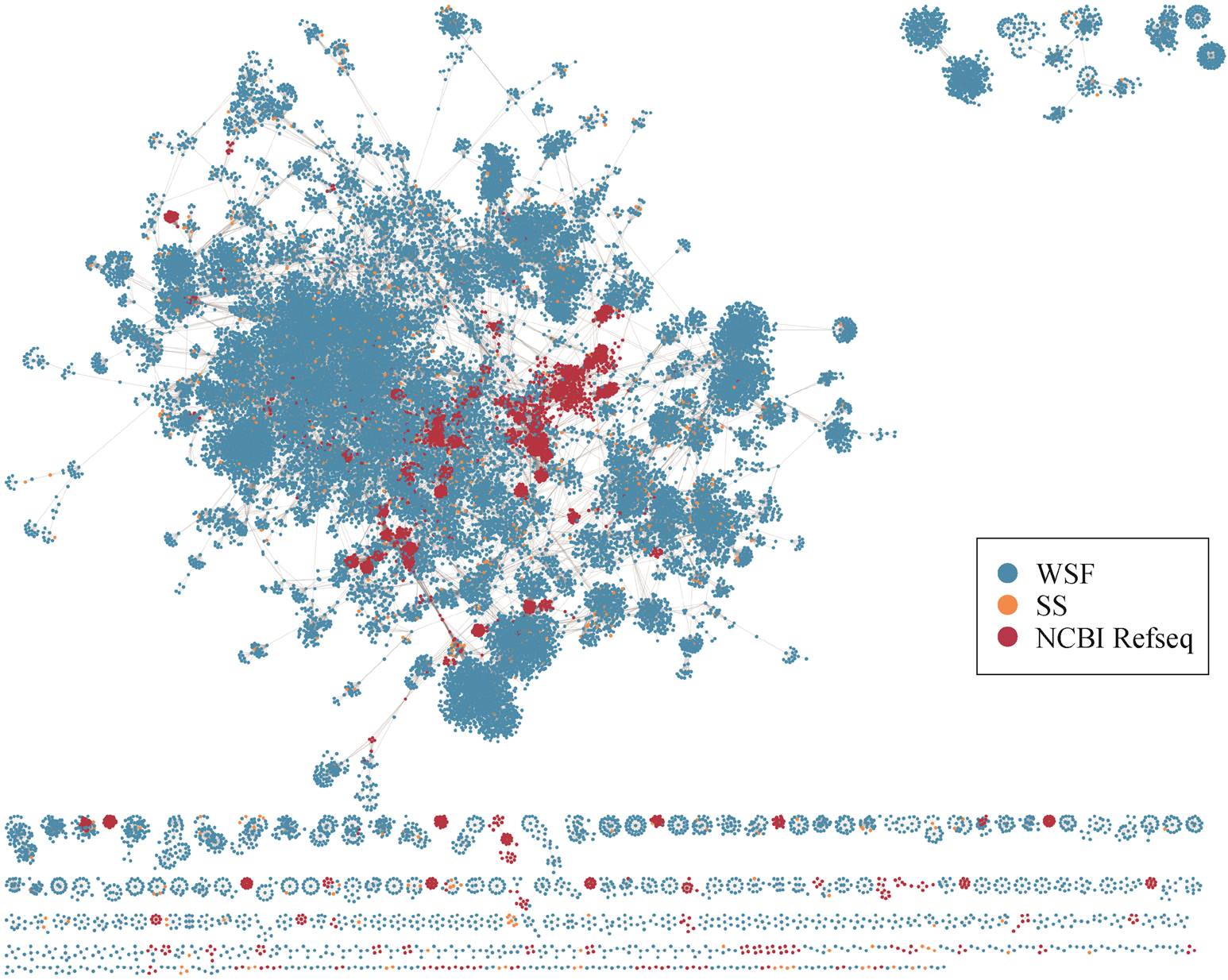
图2 黄河典型支流噬菌体群落与NCBI Refseq的共享基因网络图
Fig. 2 Gene-sharing network of phages in typical tributaries of the Yellow River and NCBI Refseq
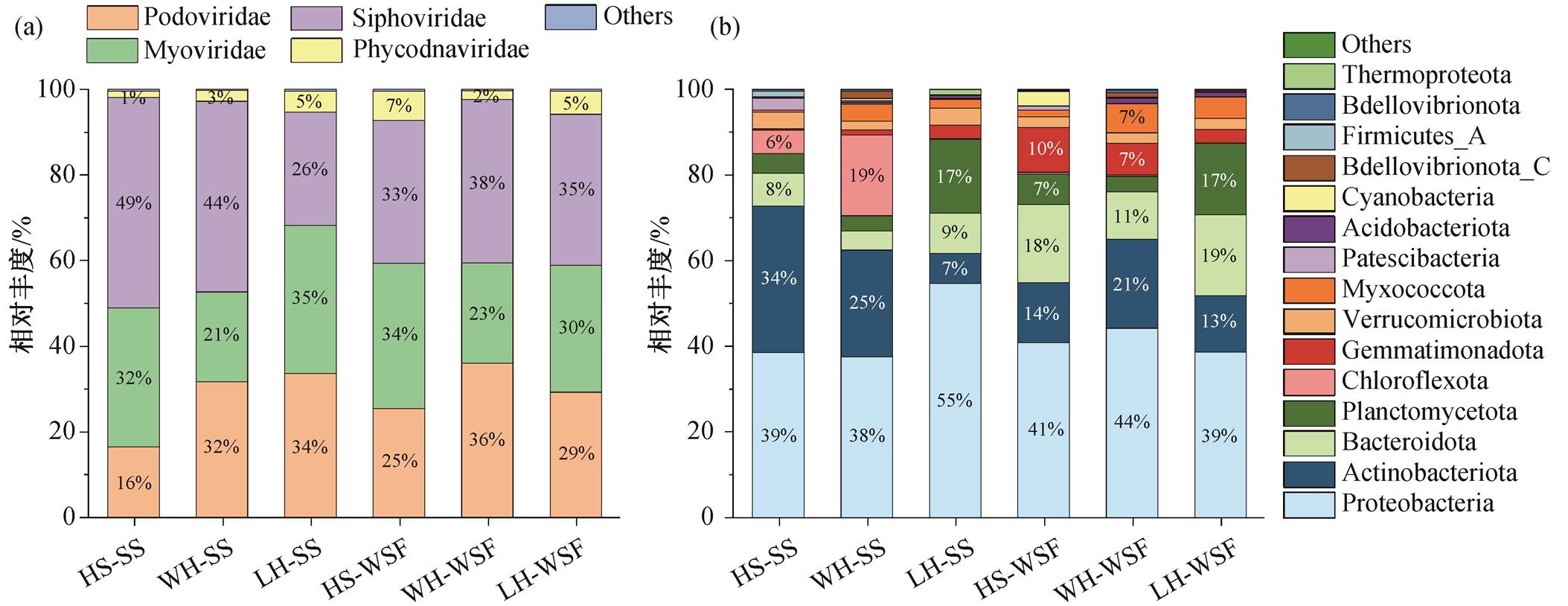
图3 不同采样区中, 按噬菌体科水平统计(a)和按宿主门水平统计(b)的vOTUs相对丰度分布
Fig. 3 The relative abundance of vOTUs in different sampling areas calculated at the level of phage families (a) and at the level of host phylum (b)
共有 2589 个 vOTUs 被预测感染来自 18 个门的原核宿主, 主要的宿主为 Proteobacteria, Actinobac-teriota, Bacteroidota, Verrucomicrobiota, Planctomy-cetota 和 Chloroflexota 等门。如图 3(b)所示, 按相对丰度统计, 感染 Proteobacteria, Actinobacteriota 和Bacteroidota 的噬菌体的相对丰度在各类样本中均超过 60%。洛河水体及沉积物中富集了感染 Planc-tomycetota 的噬菌体; 渭河沉积物样本中, 感染 Ch- loroflexota 的噬菌体相对丰度较高。
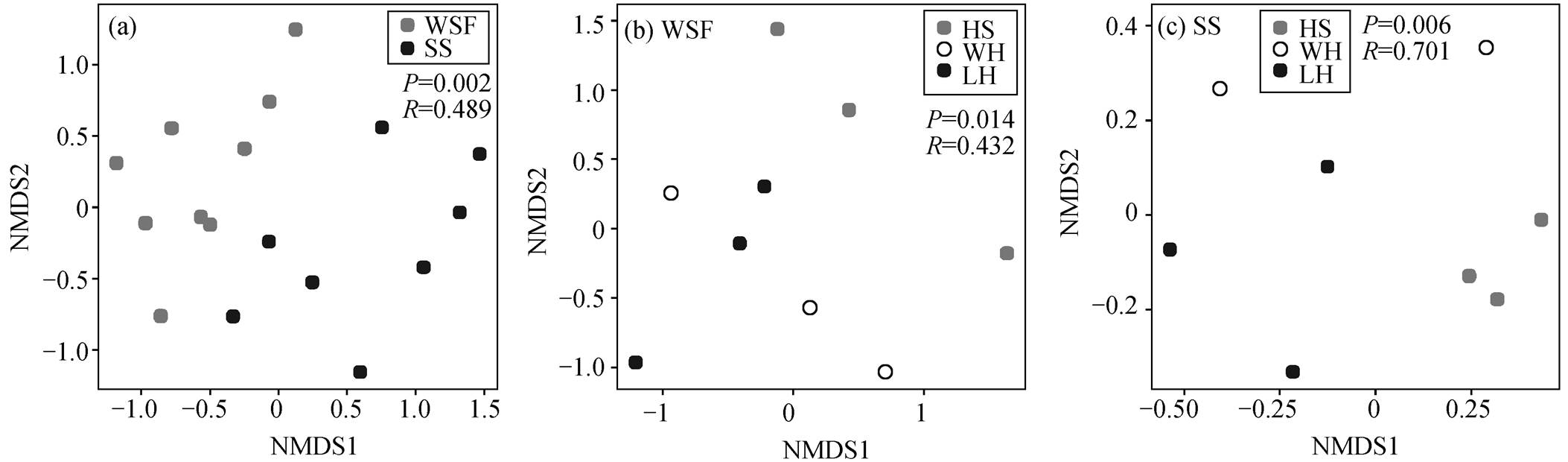
图4 黄河典型支流入干区噬菌体群落的介质和空间差异
Fig. 4 Habitat and spatial differences of phage community in confluence areas from typical tributaries to mainstream of the Yellow River
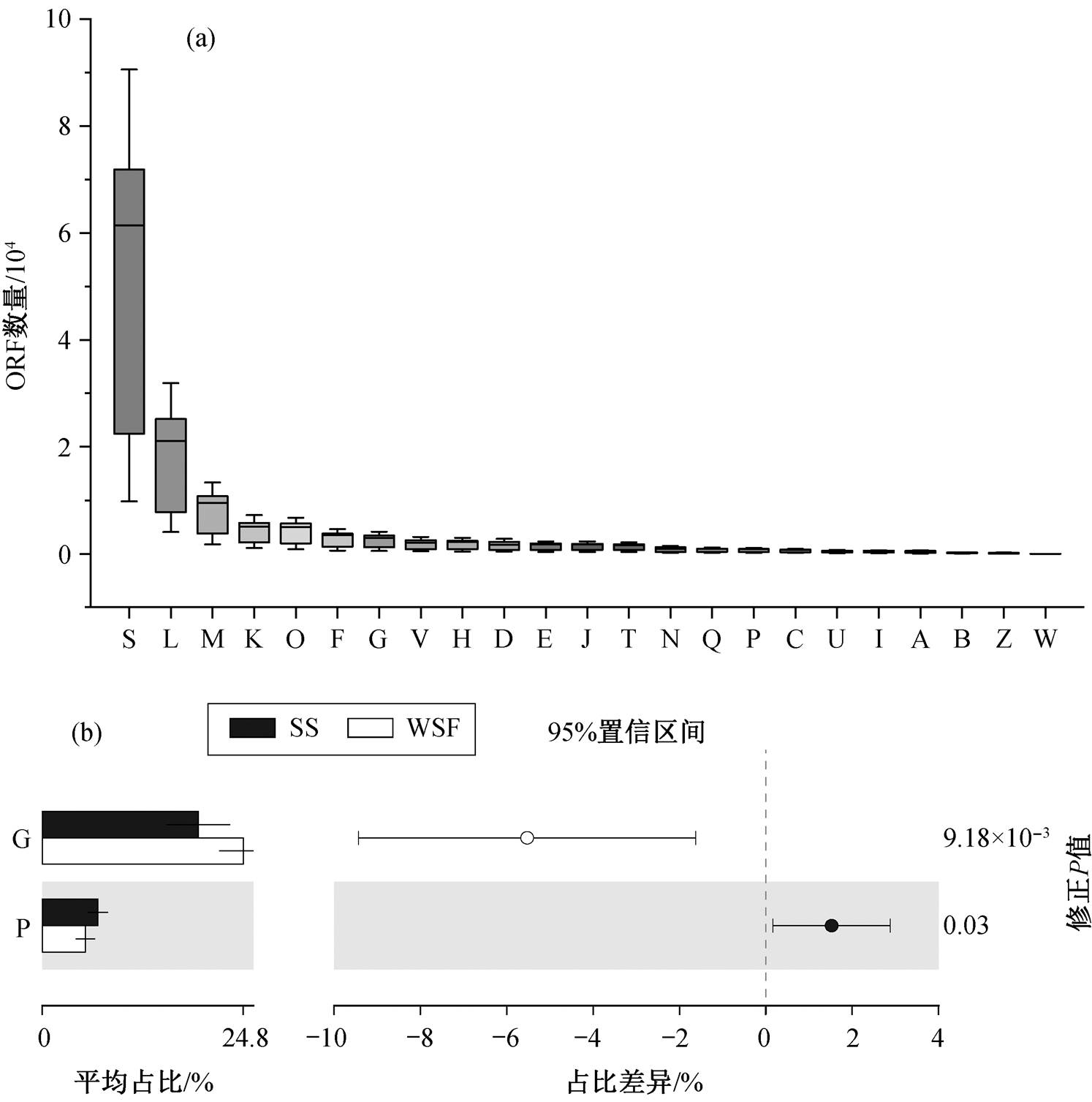
A: RNA processing and modification; B: Chromatin structure and dynamics; C: Energy production and conversion; D: Cell cycle control, cell division, chromosome partitioning; E: Amino acid transport and metabolism; F: Nucleotide transport and metabolism; G: Carbohydrate transport and metabolism; H: Coenzyme transport and metabolism; I: Lipid transport and metabolism; J: Translation, ribosomal structure and biogenesis; K: Transcription; L: Replication, recombination and repair; M: Cell wall/membrane/envelope biogenesis; N: Cell motility; O: Post-translational modification, protein turnover, and chaperones; P: Inorganic ion transport and metabolism; Q: Secondary metabolites biosynthesis, transport, and catabolism; S: Function unknown; T: Signal transduction mechanisms; U: Intracellular trafficking, secretion, and vesicular transport; V: Defense mechanisms; W: Extracellular structures; Z: Cytoskeleton
图5 噬菌体编码的功能基因种类及数量
Fig. 5 Type and number of functional genes encoded by the phage
Procrustes 分析结果(图 6(a))显示, 噬菌体群落结构与细菌宿主群落结构具有显著一致性。其中, 水体样本中二者的关联度(M2=0.17)高于沉积物样本(M2=0.49), 噬菌体与细菌宿主联系更为紧密。在门水平上进行分类时, 噬菌体的丰度与细菌宿主的丰度组成基本上一致, 噬菌体与其宿主的相对丰度显著正相关(r=0.9, p<2.2×10−16), 如图 6(b)所示。湟水河、渭河和洛河采样区的噬菌体群落中, 大部分噬菌体以微生物群落中优势物种为目标, 符合“Killing the winner”假说[43], 对水生浮游系统[44]和深海冷渗泉[45]等环境的研究结果展现出类似的规律。
46 个 vOTUs 感染超过 2 个门的细菌宿主, 占1.8%, 其中 42 个 vOTUs 能够感染 2 个细菌门, 有 4个 vOTUs 展现出感染 3 个门宿主的潜力。宿主范围与感染成功率负相关[46], 因此普遍认为噬菌体具有宿主专一性。本文结果和其他类似研究结果[47]均显示, 部分噬菌体可能存在广泛的宿主, 但该结论需要培养实验的进一步验证。
本研究调查了黄河 3 条典型支流入干区的水体和沉积物中噬菌体群落组成、功能基因的多样性以及其与宿主群落的关系, 并对支流汇入前后的干流噬菌体群落多样性变化进行研究, 得到以下认识。
1)噬菌体群落具有较高的新颖性, 99.77%的vOTUs 无法与 NCBI Refseq 参考序列聚类, 可能为未知噬菌体。有尾噬菌体目为水体和沉积物中噬菌体群落中的绝对优势类群, 相对丰度超过 90%。
2)在检出的隶属于 15 科、158 属的噬菌体中, 湟水河中特有噬菌体的属最多, 如 Pbi1virus, Bad-navirus, Ea214virus 和 Gammabaculovirus 等。湟水河、渭河及洛河采样区域的噬菌体群落组成及结构存在差异, 且在 3 条支流的沉积物中噬菌体的群落差异大于水体, 空间异质性作用更强。同时, 水体和沉积物样本中噬菌体群落结构具有显著差异, 存在生境异质性。
3)噬菌体群落编码的功能蛋白主要维持其繁殖和转录, AMGs 主要参与核酸、碳水化合物和氨基酸的代谢和转运过程, 对湟水河、渭河及洛河采样区域的氮、碳和磷等元素的循环过程具有潜在影响。AMGs 的分布具有介质差异, 水体群落富集碳水化合物的运输和代谢类基因, 而沉积物群落富集无机离子的运输和代谢类基因。
4)噬菌体群落的宿主隶属 18 个门的原核生物, 洛河的水体及沉积物中富集感染 Planctomycetota 的噬菌体; 渭河沉积物样本中, 感染 Chloroflexota的噬菌体相对丰度较高。约有 1.8%的vOTUs 具有感染 2 个门及以上宿主的潜在能力。在门水平上, 噬菌体群落结构与细菌宿主群落结构具有显著的一致性, 且在水体中二者的关联度更高。
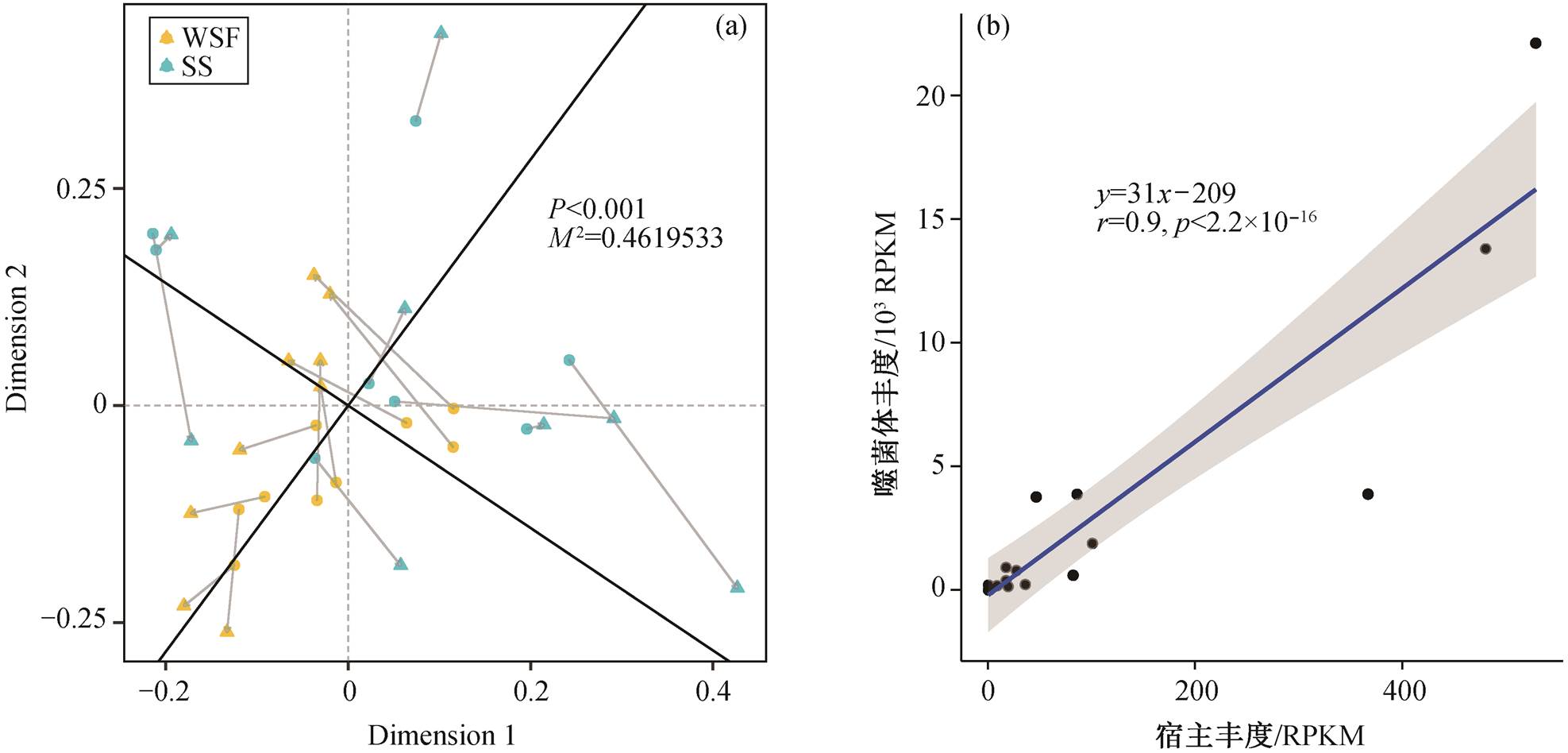
图6 噬菌体群落与细菌群落的相关性
Fig. 6 Correlation between phage community and bacteria community
参考文献
[1] Dion M B, Oechslin F, Moineau S. Phage diversity, genomics and phylogeny. Nat Rev Microbiol, 2020, 18(3): 125–138
[2] Suttle C A. Marine viruses-major players in the global ecosystem. Nat Rev Microbiol, 2007, 5(10): 801–812
[3] Zhang Y Z, Shi M, Holmes E C. Using metagenomics to characterize an expanding virosphere. Cell, 2018, 172(6): 1168–1172
[4] Roux S, Brum J R, Dutilh B E, et al. Ecogenomics and potential biogeochemical impacts of globally abundant ocean viruses. Nature, 2016, 537: 689–693
[5] Gregory A C, Zayed A A, Conceição-Neto N, et al. Marine DNA viral macro- and microdiversity from pole to pole. Cell, 2019, 177(5): 1109–1123
[6] Jin M, Guo X, Zhang R, et al. Diversities and potential biogeochemical impacts of mangrove soil viruses. Microbiome, 2019, 7(1): 58
[7] Zheng X, Jahn M T, Sun M, et al. Organochlorine contamination enriches virus-encoded metabolism and pesticide degradation associated auxiliary genes in soil microbiomes. The ISME Journal, 2022, 16(5): 1397–1408
[8] Kavagutti V S, Andrei A, Mehrshad M, et al. Phage-centric ecological interactions in aquatic ecosystems revealed through ultra-deep metagenomics. Microbio-me, 2019, 7(1): 135
[9] 周莘皓, 梁彦韬, McMinn A, 等. 极地海洋病毒宏基因组研究进展. 极地研究, 2021, 33(4): 612–620
[10] Camarillo-Guerrero L F, Almeida A, Rangel-Pineros G, et al. Massive expansion of human gut bacterio-phage diversity. Cell, 2021, 184(4): 1098–1109
[11] Roux S, Adriaenssens E M, Dutilh B E, et al. Mini-mum information about an uncultivated virus genome (MIUViG). Nat Biotechnol, 2019, 37(1): 29–37
[12] Camargo A P, Nayfach S, Chen I A, et al. IMG/VR v4: an expanded database of uncultivated virus genomes within a framework of extensive functional, taxono-mic, and ecological metadata. Nucleic Acids Research, 2023, 51(D1): D733–D743
[13] Paez-Espino D, Eloe-Fadrosh E A, Pavlopoulos G A, et al. Uncovering Earth’s virome. Nature, 2016, 536: 425–430
[14] Jarett J K, Džunková M, Schulz F, et al. Insights into the dynamics between viruses and their hosts in a hot spring microbial mat. The ISME Journal, 2020, 14(10): 2527–2541
[15] Gazitúa M C, Vik D R, Roux S, et al. Potential virus-mediated nitrogen cycling in oxygen-depleted oceanic waters. The ISME Journal, 2021, 15(4): 981–998
[16] Chen L, Méheust R, Crits-Christoph A, et al. Large freshwater phages with the potential to augment aero-bic methane oxidation. Nat Microbiol, 2020, 5 (12): 1504–1515
[17] Chen Y, Wang Y, Paez-Espino D, et al. Prokaryotic viruses impact functional microorganisms in nutrient removal and carbon cycle in wastewater treatment plants. Nat Commun, 2021, 12(1): 5938
[18] 李玉红. 黄河流域干支流水污染治理研究. 经济问题, 2021(5): 9–15
[19] 厉治平, 穆航, 樊晶晶. 渭河支流汇入对干流径流变异的影响分析. 海河水利, 2019(6): 25–28
[20] 徐火清, 赵红红, 吴义军. 金沙江下游主要支流对干流水温的影响分析. 水利水电科技进展, 2023, 43(1): 22–28
[21] Zhao E, Kuo Y, Chen N. Assessment of water quality under various environmental features using a site-specific weighting water quality index. Sci Total En-viron, 2021, 783(5): 146868
[22] Ding Y, Li M, Pan B, et al. Disentangling the drivers of phytoplankton community composition in a heavily sediment-laden transcontinental river. J Environ Ma-nage, 2022, 302(Pt.A): 113939
[23] Pan B, Liu X, Sun H, et al. Suspended particulates mediate bacterial community coalescence in different habitats of a large sediment-laden river. Ecol Indic, 2022, 144(4): 109462
[24] Yu Q, Yang J, Su W, et al. Heavy metals and micro-biome are negligible drivers than mobile genetic ele-ments in determining particle-attached and free-living resistomes in the Yellow River. J Hazard Mater, 2022, 424: 127564
[25] He H, Pan B, Yu K, et al. Determinants of bacte-rioplankton structures in the typically turbid Weihe River and its clear tributaries from the northern foot of the Qinling Mountains. Ecol Indic, 2021, 121: 107168
[26] 刘峰, 冯民权, 王毅博. 汾河入黄口夏季微生物群落结构分析. 微生物学通报, 2019, 46(1): 54–64
[27] Uritskiy G V, Diruggiero J, Taylor J. MetaWRAP — a flexible pipeline for genome-resolved metagenomic data analysis. Microbiome, 2018, 6(1): 158
[28] Nayfach S, Camargo A P, Schulz F, et al. CheckV assesses the quality and completeness of metagenome-assembled viral genomes. Nat Biotechnol, 2021, 39(5): 578–585
[29] Fu L, Niu B, Zhu Z, et al. CD-HIT: accelerated for clustering the next-generation sequencing data. Bioin-formatics, 2012, 28(23): 3150–3152
[30] Pons J C, Paez-Espino D, Riera G, et al. VPF-Class: taxonomic assignment and host prediction of uncul-tivated viruses based on viral protein families. Bioin-formatics, 2021, 37(13): 1805–1813
[31] Hyatt D, Chen G, Locascio P F, et al. Prodigal: proka-ryotic gene recognition and translation initiation site identification. Bmc Bioinformatics, 2010, 11(1): 119
[32] Huerta-Cepas J, Szklarczyk D, Heller D, et al. eggNOG 5.0: a hierarchical, functionally and phylogenetically annotated orthology resource based on 5090 organisms and 2502 viruses. Nucleic Acids Research, 2019, 47 (D1): D309–D314
[33] Chaumeil P, Mussig A J, Hugenholtz P, et al. GTDB-Tk: a toolkit to classify genomes with the Geno- me Taxonomy Database. Bioinformatics, 2019, 36(6): 1925–1927
[34] Langmead B, Salzberg S L. Fast gapped-read align-ment with Bowtie 2. Nat Methods, 2012, 9(4): 357–359
[35] Li H, Handsaker B, Wysoker A, et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics, 2009, 25(16): 2078–2079
[36] Tominaga K, Morimoto D, Nishimura Y, et al. In silico prediction of virus-host interactions for marine bac-teroidetes with the use of metagenome-assembled genomes. Front Microbiol, 2020, 11: 738
[37] 徐步, 邹雪蓉, 朱元清, 等. 环境病毒的宿主鉴定技术进展. 微生物学报, 2022, 62(12): 4663–4683
[38] 赵梦瑶, 梁恩航, 陈颖, 等. 黄河玛曲至临河段硅藻群落组成及水质评价. 北京大学学报(自然科学版), 2022, 58(1): 169–176
[39] 张奇亚. 淡水生态系统中几种大DNA病毒研究概述. 水生生物学报, 2020, 44(5): 961–975
[40] 吴雷祥, 王卓微, 黄伟, 等. 河流连续体理论研究进展// 2021第九届中国水生态大会. 西安, 2021: 1–10
[41] 陈焰, 夏瑞, 后希康. 黄河上游流域水生态环境问题及对策——以湟水河典型流域为例. 环境保护, 2021, 49(13): 17–19
[42] 贾杰, 姜丽红. 渭河源区输沙量演变特征及其影响因素分析. 水电能源科学, 2022, 40(8): 45–48
[43] Bouvier T, Del Giorgio P A. Key role of selective viral-induced mortality in determining marine bacterial community composition. Environmental Microbiolo-gy, 2007, 9(2): 287–297
[44] Brussaard C P D. Viral control of phytoplankton popu-lations — a review. The Journal of Eukaryotic Mic-robiology, 2004, 51(2): 125–138
[45] Li Z, Pan D, Wei G, et al. Deep sea sediments associa-ted with cold seeps are a subsurface reservoir of viral diversity. The ISME Journal, 2021, 15: 2366–2378
[46] Peters D L, Lynch K H, Stothard P, et al. The isolation and characterization of two Stenotrophomonas malto-philia bacteriophages capable of cross-taxonomic or-der infectivity. BMC Genomics, 2015, 16(1): 664
[47] Schulz F, Roux S, Paez-Espino D, et al. Giant virus diversity and host interactions through global meta-genomics. Nature, 2020, 578: 432–436
Characterization of the Phage Community Structure in Typical Tributaries of the Yellow River
Abstract Metagenomics was used to analyze the phage community structure in the water and sediment of Huangshui River, Weihe River and Luohe River, and the diversity, host and functional potential of the three tributaries of the Yellow River were compared. The results showed that 99.77% of the unknown vOTUs (viral Operational Taxonomic Units) identified from the metagenome could not be clustered with the NCBI Refseq database, indicating a high degree of species novelty in the phage community. A total of 158 genera belonging to 15 families were detected in three tributaries. The endemic phage genera were the most in Huangshui River and Pbi1virus, Badnavirus, Ea214virus and Gammabaculovirus were detected only in this tributary. The phage community structure of the three tributaries was significantly different and the spatial heterogeneity was greater in the sediment samples. Diversified auxiliary metabolic genes (AMGs) indicated that phages might be involved in the cycling of nitrogen, carbon and phosphorus in Huangshui River, Weihe River and Luohe River, but there was no difference in the distribution of AMGs in the three tributaries. The host range of phage community spanned 18 prokaryotic phyla, mainly including Proteobacteria, Actinobacteriota and Bacteroidota. About 1.8% of the phages might infect hosts across phyla. Phages infecting Planctomycetota were enriched in water and sediments of Luohe River, while Chloroflexota-infected phages were relatively abundant in the sediments of Weihe River. There was a significant correlation between the bacterial and phage community and the consistency was higher in water samples.
Key words tributary; viral metagenomics; phage community; community structure