图1 研究区域采样点
Fig. 1 Sampling sites in the study area
北京大学学报(自然科学版) 第59卷 第1期 2023年1月
Acta Scientiarum Naturalium Universitatis Pekinensis, Vol. 59, No. 1 (Jan. 2023)
doi: 10.13209/j.0479-8023.2022.124
国家自然科学基金(41907203)资助
收稿日期: 2022–03–02;
修回日期: 2022–04-06
摘要 基于 2014 年 3 月和 10 月汉江中下游 6 个断面的水体和沉积物监测样品, 获得 384 个高质量 vOTUs (viral Operational Taxonomic Units)。研究表明, vOTUs 中 95%以上隶属 Caudovirales (有尾噬菌体目), 丰度居前 3 位的科分别为 Myoviridae (肌尾噬菌体科)、Siphoviridae (长尾噬菌体科)和 Podoviridae(短尾噬菌体科)。主坐标分析和相似性分析结果表明, 沉积物中病毒群落结构相对稳定, 而水体中群落易随季节发生变化, 同流域的水体和沉积物中群落可能具有连通性。宿主来自 2 域(界) 19 门, 丰度最高的宿主是 Proteobacteria 门(变形菌门)。88%的 vOTUs 具有单一的门水平的宿主, 有 3 个 vOTUs 跨越 5 个门。跨域(界)感染的 vOTUs 在感染古菌界 Thermoproteota 门的同时, 最常见的是感染细菌界的 Bacteroidota 门(拟杆菌门)。与水体相比, 沉积物中宿主的群落组成更加多样。Pearson 相关分析表明, 噬菌体与其宿主的组成在门水平上一致。
关键词 汉江; 噬菌体; 宿主; 水体; 沉积物
自然界中, 病毒无处不在。病毒是一类无法自行表现出生命现象、不能离开宿主细胞独立进行繁殖和代谢的类生物, 是介于生命体与非生命体之间的有机物种。噬菌体是一类以细菌等为宿主的病毒, 在生物地球化学循环中起着重要作用, 在水生环境中, 每天杀死海洋中近 20%的生物质, 使碳流短路[1],影响物质和能量循环[2–3]。噬菌体能够通过水平基因转移和调控宿主的微生物种群, 调节微生物群落结构, 它们在生物群落之间积极移动, 是进化的主要推动者[4]。噬菌体还是未经开发的遗传多样性的最大储库之一。
基于传统的分离培养、整合宏基因组学[5]和单基因组学[6]等方法, 能够了解病毒与宿主间的相互作用, 却不够全面和详尽。宏基因组学能够针对大量未培养的噬菌体病毒构建大规模的数据集, 刻画其群落结构。在对水体的研究中, 对太平洋[7]、马拉皮纳海洋[8]和塔拉海洋[9]的研究都在大尺度范围内, 对来自不同深度和位置的海洋样本进行分析, 刻画海洋中双链 DNA 病毒的局部和全球尺度的丰度模式[10]。此外, 有研究从 3042 个不同地理起源的宏基因组中检测到大量病毒的基因组[11], 同时发现在全球范围内, 尽管在海洋和人类样品中的发现率可能已经接近饱和, 但总的基因组多样性仍未得到广泛的表征。在任何给定的环境样品序列中, 大多数序列仍是未知的, 有时未知序列占 reads 结果的 90%以上[2]。目前, 国外几项重要研究对海洋中的病毒进行了详尽的报道, 但对淡水系统中噬菌体多样性的认知仍然有限。国内对淡水中病毒的研究也较少[12], 仅对武汉东湖[13]、福建九龙江河口[14]和高原湖泊[15]等有个别的研究。
汉江是长江第一大支流, 发源于陕西省秦岭南麓, 在武汉龙王庙汇入长江。本文选取汉江具有代表性的河段进行采样, 了解淡水系统水体和沉积物中噬菌体的特征, 刻画其与宿主群落的丰度关系。
参照长江水利委员会水文局设置的长江流域国家水文监测站的位置, 设置从上游到下游的 1~6 号采样点, 分别为白河、丹江口坝下、陶岔、襄阳、仙桃和集家嘴(图 1), 于 2014 年 3 月(春季)和 10 月(秋季)采集水体和沉积物样品。样品按照季节和介质类型标记为 WS (Water_Spring, 春季水体)、WA (Water_Autumn, 秋季水体)、SS (Sediment_Spring, 春季沉积物)和 SA (Sediment_Autumn, 秋季沉积物)。其中, 春季沉积物的 1 号和 3 号采样点样品在多次提取 DNA 后未达宏基因组建库标准, 因而缺失后续数据。
水样采集采用等比例混合水样采集法, 在每个采样断面采集 10~15L 的水样, 在 24 小时之内使用0.22μm 聚碳酸酯滤膜(Millipore, USA)过滤, 保存滤膜。沉积物样品的采集使用抓斗式采泥器, 在水样采集点下方抓取表层沉积物。
水体样品使用 FastDNA® SPIN Kit for Soil(MP Biomedicals, 美国)试剂盒提取 DNA, 沉积物样品离心后取样, 用相同方式提取。每个环境样本进行3~5 次 DNA 抽提, 将每个环境样本的 3~5 个 DNA样品混合。使用 1%的琼脂糖凝胶电泳检测 DNA 完整性, 用 Nanodrop 2000 (Thermo Scientific, 美国)进行定量分析。然后用干冰保存, 送至上海美吉生物医药科技有限公司, 使用 Illumina Hiseq 4000 平台(Illumina, 美国)进行双端的宏基因组测序。
使用 TrimGalore v0.6.4[16]去除低质量条带, 然后使用 metaWRAP v1.2.2[17]中的 metaspades 进行拼接。病毒识别参照 Gregory 等[18]的方法, 用 CheckV评估质量和完整性[19], 筛选并设置长度阈值为 3kb, 得到假定的序列。使用 CD-HIT[20]聚类去冗余后得到 vOTUs。
图1 研究区域采样点
Fig. 1 Sampling sites in the study area
基因共享网络的构建首先使用 prodigal[21]预测蛋白, 然后使用 vConTACT2[22], 将蛋白与 NCBI Refseq (NCBI Reference Sequence Database)[23]数据库进行比对。
丰度计算参考 Mende 等[24]的方法, 使用 bow-tie2[25]及 samtools[26], 计算得到 RPKM 模式(每千碱基 reads 映射到的 reads)归一化后的丰度表, 物种注释使用 VPF-tools[27]。
使用 mataWRAP[17]对宿主装箱, 对所有 contigs取完整度为 50, 污染率为 10%的筛选值。用 GTDB- tk[28]获得宿主物种注释。最后, 比对病毒与宿主的bins[29], 进行宿主预测。宿主的丰度取所在 bins 的丰度, 丰度计算方法与病毒相同。
利用 Cytoscape[30], 对 vConTACT2 聚类结果进行网络可视化。数据可视化使用 R (v4.0.0)软件的ggplot2 包, 使用 VennDiagram 包绘制韦恩图, 使用pheatmap 包绘制热图。主坐标分析(principal coor-dinates analysis, PCoA)和相似性分析(analysis of similarities, ANOSIM)使用 vegan 包计算 Bray-Curtis距离。
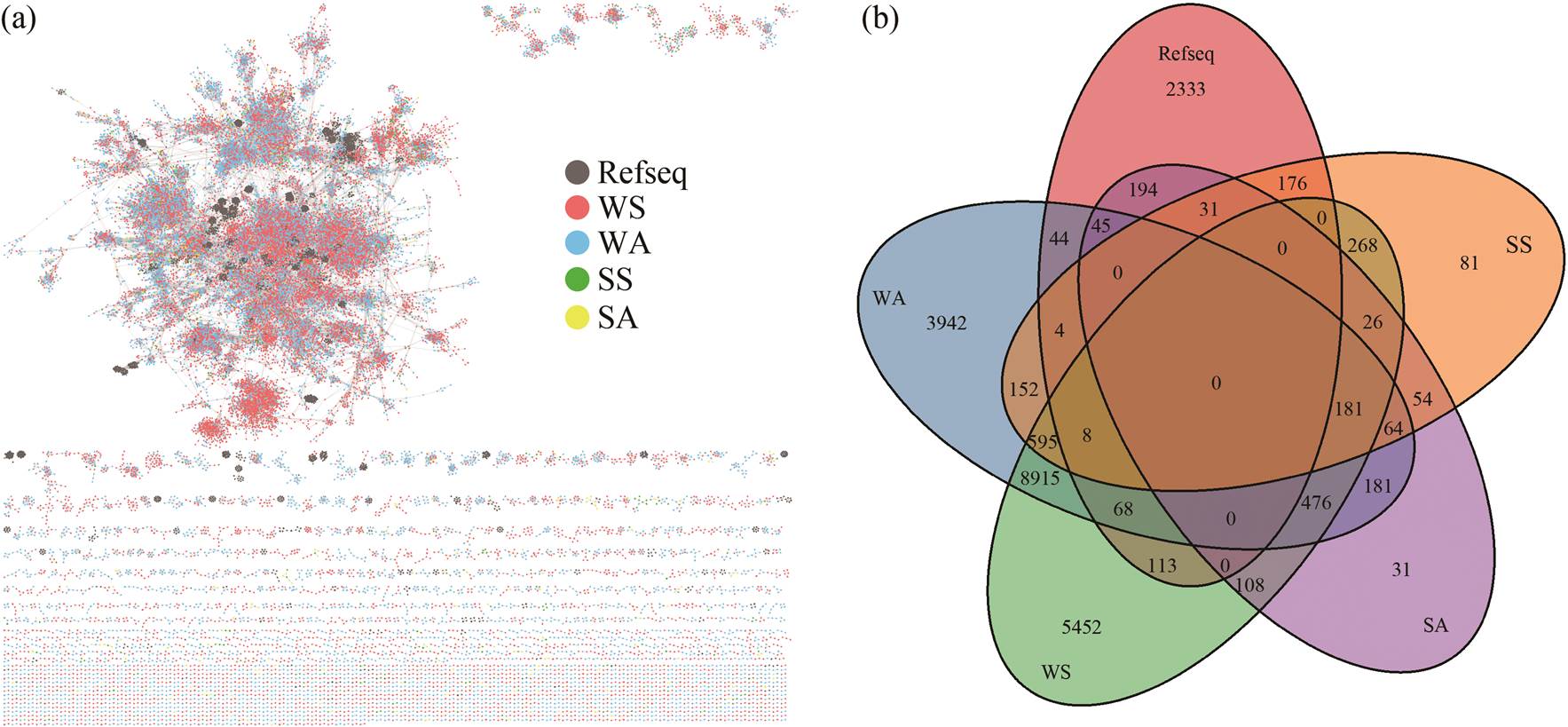
WA: 秋季水体; WS: 春季水体; SA: 秋季沉积物; SS: 春季沉积物
图2 vOTUs与Refseq数据库的聚类网络(a)和共享数量的韦恩图(b)
Fig. 2 Cluster network of vOTUs with Refseq (a) and Venn diagram of shared numbers (b)
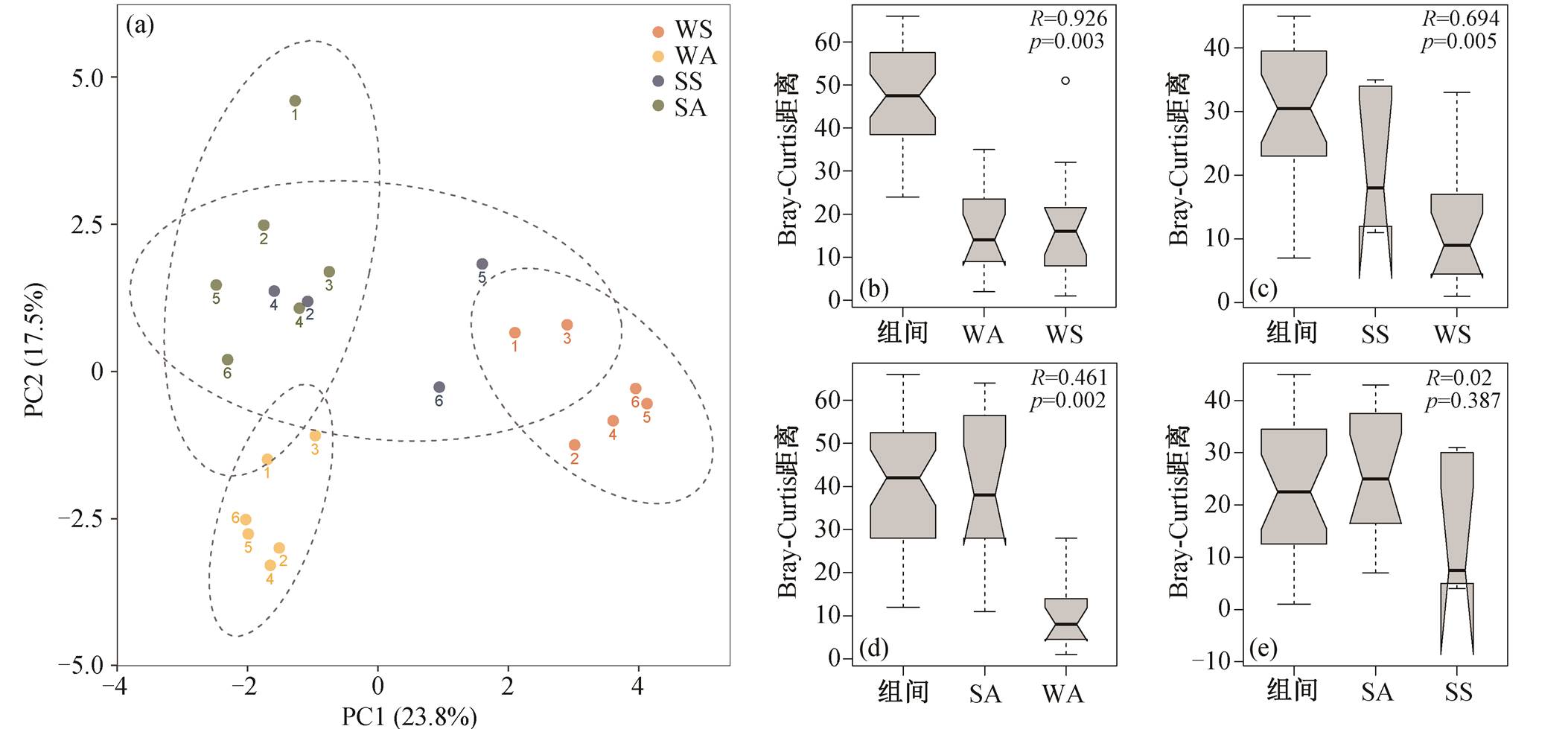
图3 汉江水体和沉积物中vOTUs的PCoA分析(a)和分组的ANOSIM分析((b)~(e))
Fig. 3 PCoA (a) and grouped ANOSIM ((b)–(e)) analysis of vOTUs in water and sediments of Hanjiang River
在所有样点中得到假定的病毒序列 163274 条, 其中 CheckV 评估为完整质量的序列 133 条, 高质量的序列 323 条, 中等质量的序列 969 条, 低质量的序列 161849 条。聚类后得到非冗余的 109832 个vOTUs, 去冗余率约为 33%。
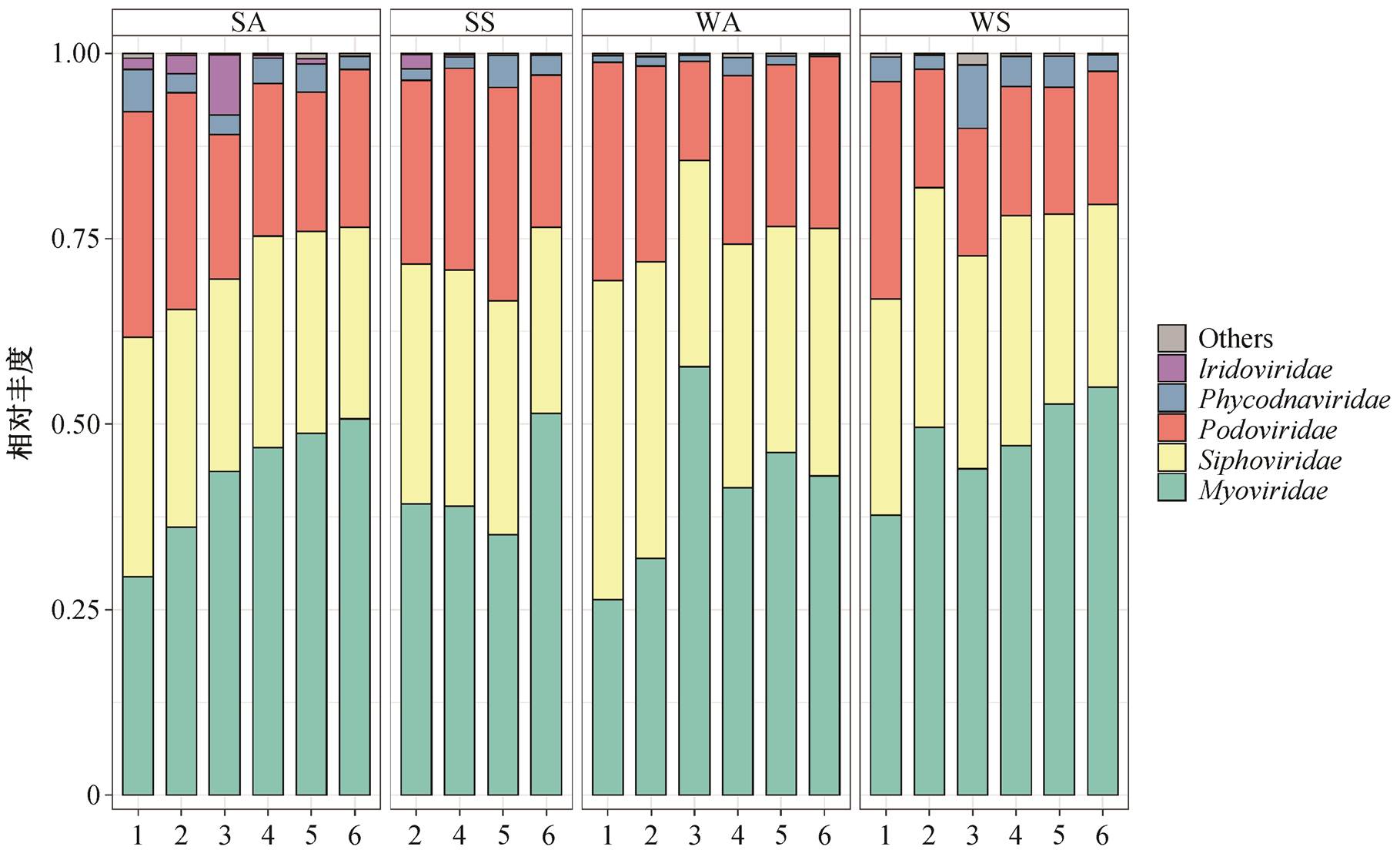
图4 科水平病毒的丰度分布
Fig. 4 Abundance distribution of viruses of family level
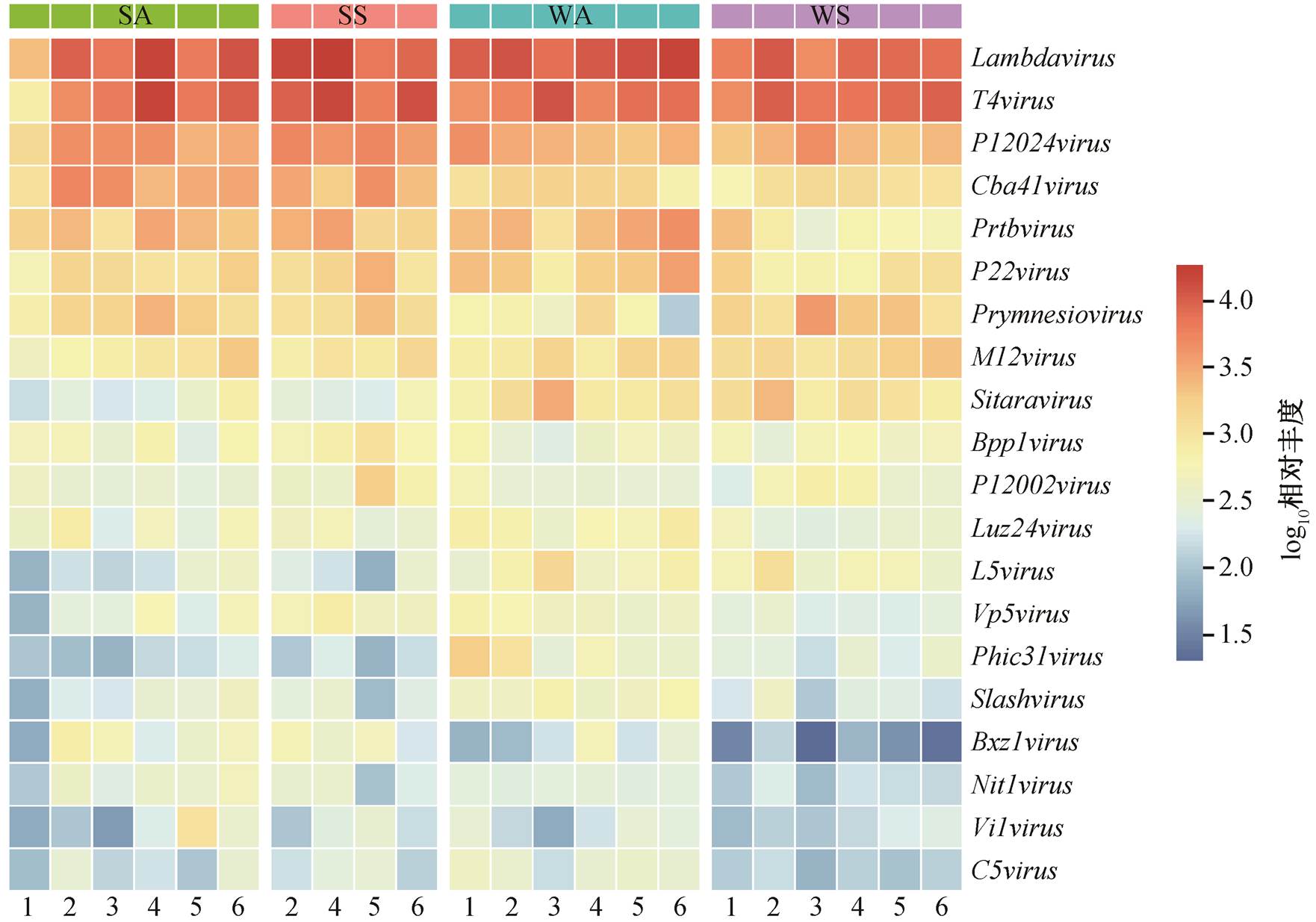
图5 属水平相对丰度排名前20的热图
Fig. 5 Heatmap depiction of relative abundance of the top 20 genera
Refseq 数据库保存了较为可靠的高质量序列, 将 vOTUs 与该库比对, 构建蛋白共享网络, 在网络中的簇能达到接近属水平的分类。如图 2(a)所示, 序列与 3503 条来自 Refseq 数据库的 contigs 聚类形成 7496 个簇, 总体聚类较松散。图 2(b)显示不同季节的水体样品具有最多的能够聚类的序列, 体现出在属水平上极大的相似度和关联性, 而春秋季的沉积物样品在属水平上的相似度不高。只有 181 个vOTUs 在所有的四类生态类型中共享, 占比不到3%, 没有簇在全部 5 类系统中都存在, 体现出汉江中噬菌体的特异性。
对聚类后的 vOTUs 进行物种注释, 在科水平注释得到 28891 个 vOTUs, 注释率为 26.3%, 分属 13个科。其中 Myoviridae (肌尾噬菌体科)注释得到13648 个 vOTUs, Siphoviridae (长尾噬菌体科)注释到 7744 个 vOTUs, Podoviridae (短尾噬菌体科)注释到 6114 个 vOTUs。此外, Phycodnaviridae 科注释到1176 个 vOTUs, Mimiviridae (拟菌病毒科)注释到154 个 vOTUs, Lavidaviridae (大病毒依赖科)注释到19 个 vOTUs, Iridoviridae 科注释到 18 个 vOTUs, Microviridae (微小噬菌体科)注释到 11 个 vOTUs。另外, Inoviridae (丝状噬菌体科)和 Adenoviridae (腺病毒科)各注释到 2 个 vOTUs, Marseilleviridae (马赛病毒科)和 Poxviridae (痘病毒科)各注释到 1 个vOTUs。肌尾、长尾和短尾噬菌体科病毒具有 95%以上的压倒性占比, 很大程度上是由于 SEA-PHAGES 计划[31]对 1537 个长尾噬菌体进行了分离和基因组测序, 导致公共基因组数据库中目前最丰富的噬菌体目是有尾噬菌体目, 随着新病毒的发现, 未来有尾噬菌体不成比例的表现可能会减少[2]。
在属水平, 注释出 18109 个 vOTUs, 注释率为16.5%, 分属 123 个属。总 vOTU 数占比大于 1%的优势属有 12 个, 分别为 T4virus, Lambdavirus,P12024virus, Prymnesiovirus, P22virus, Cba41virus, M12virus, Prtbvirus, Sitaravirus, Bpp1virus, P12002virus 和 Luz24virus。这 12 个优势属的总多度达到总数的87.5%。
如图 3 所示, 使用 Bray-Curtis 距离, 对 vOTUs进行 PCoA 和 ANOSIM 分析, 研究空间介质和季节变化对群落分布的影响。分组 ANOSIM 分析显示, 唯一没有显著性差异的是春季沉积物和秋季沉积物, 春季水体和秋季水体的差异最显著, 体现沉积物介质中噬菌体群落的稳定性, 而水体中的噬菌体群落易随季节发生变化。此外, 相比不同季节, 同一季节的水体与沉积物样品之间的聚类更加紧密, 体现了同流域水体和沉积物噬菌体群落的连通性。
图 4 显示科水平物种组成分布的季节变化和空间介质的差异。秋季底泥中, 从上游到下游, 肌尾噬菌体科丰度占比增加。藻类 DNA 病毒科在春季水体中的占比整体上高于秋季水体, 可能是受到宿主丰度的影响。虹彩病毒科的病毒多样性不高, 总体丰度却较高, 且沉积物样品中的丰度高于水体样品, 可能是由于虹彩病毒科主要感染鱼类、两栖类和爬行类动物等[32], 在沉积物中的宿主比水体中的更丰富。在 3 号样点, 秋季水体样品中肌尾噬菌体丰度占比明显偏高, 短尾噬菌体占比偏低, 秋季沉积物样品虹彩病毒科占比明显偏高, 春季水体样品中藻类 DNA 病毒科占比明显偏高, 可能是由于该点所处支流与汉江干流存在一定的偏离, 宿主群落组成有较大的变化。
对属水平的病毒进行分析, 结果如图 5 所示。Sitaravirus 属、L5virus 属和 Phic31virus 属在底泥中的丰度比水体中低, Cba41virus 属和 Bxz1virus 属在底泥中的丰度比水体中高。在 3 号样点, 春季水体中的 prtbvirus 属和秋季水体中的 Prymnesiovirus 属的丰度明显偏低。
共计 3140 个 vOTUs 注释到来自 2 域(界) 19 门的宿主, 注释率为 2.9%。宿主包括古菌门水平的 3个门 4 个纲(Thermoproteota 门下的 Nitrososphaeria纲和 Bathyarchaeia 纲、Thermoplasmatota 门下的 E2纲、Halobacteriota 门下的 Methanosarcinia 纲)以及细菌门水平的 16 个门, 其中主要的门(占比>1%)有 9 个: Proteobacteria, Actinobacteriota, Bacteroidota, Patescibacteria, Verrucomicrobiota, Acidobacteriota, Planctomycetota, Nitrospirota 和 Chloroflexota。其他7 个细菌门包括 Gemmatimonadota, Cyanobacteria, Armatimonadota, Myxococcota, Desulfobacterota, Ei-senbacteria和Bdellovibrionota_C。
表1 跨域(界)感染的宿主门
Table 1 Host ranges across domain (phylum)
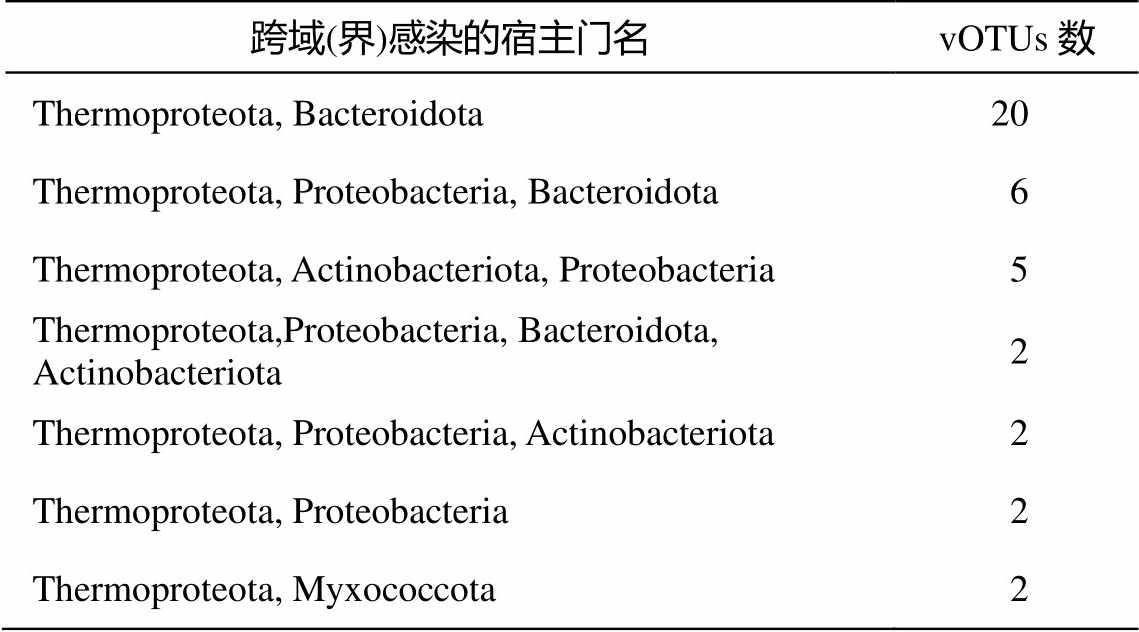
跨域(界)感染的宿主门名vOTUs数 Thermoproteota, Bacteroidota20 Thermoproteota, Proteobacteria, Bacteroidota6 Thermoproteota, Actinobacteriota, Proteobacteria5 Thermoproteota,Proteobacteria, Bacteroidota, Actinobacteriota2 Thermoproteota, Proteobacteria, Actinobacteriota2 Thermoproteota, Proteobacteria2 Thermoproteota, Myxococcota2
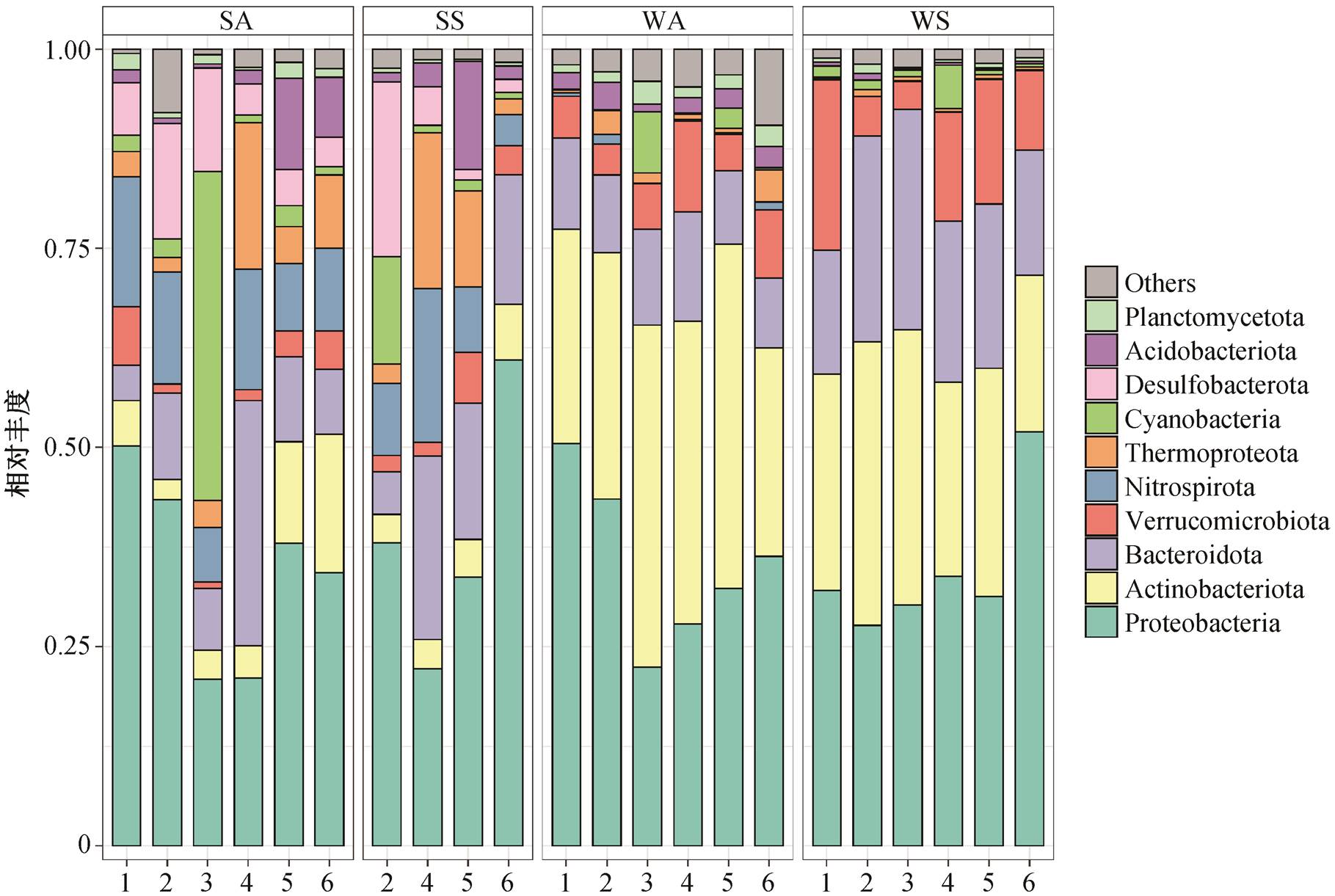
图6 门水平宿主的丰度分布
Fig. 6 Abundance distribution of hosts of phylum level
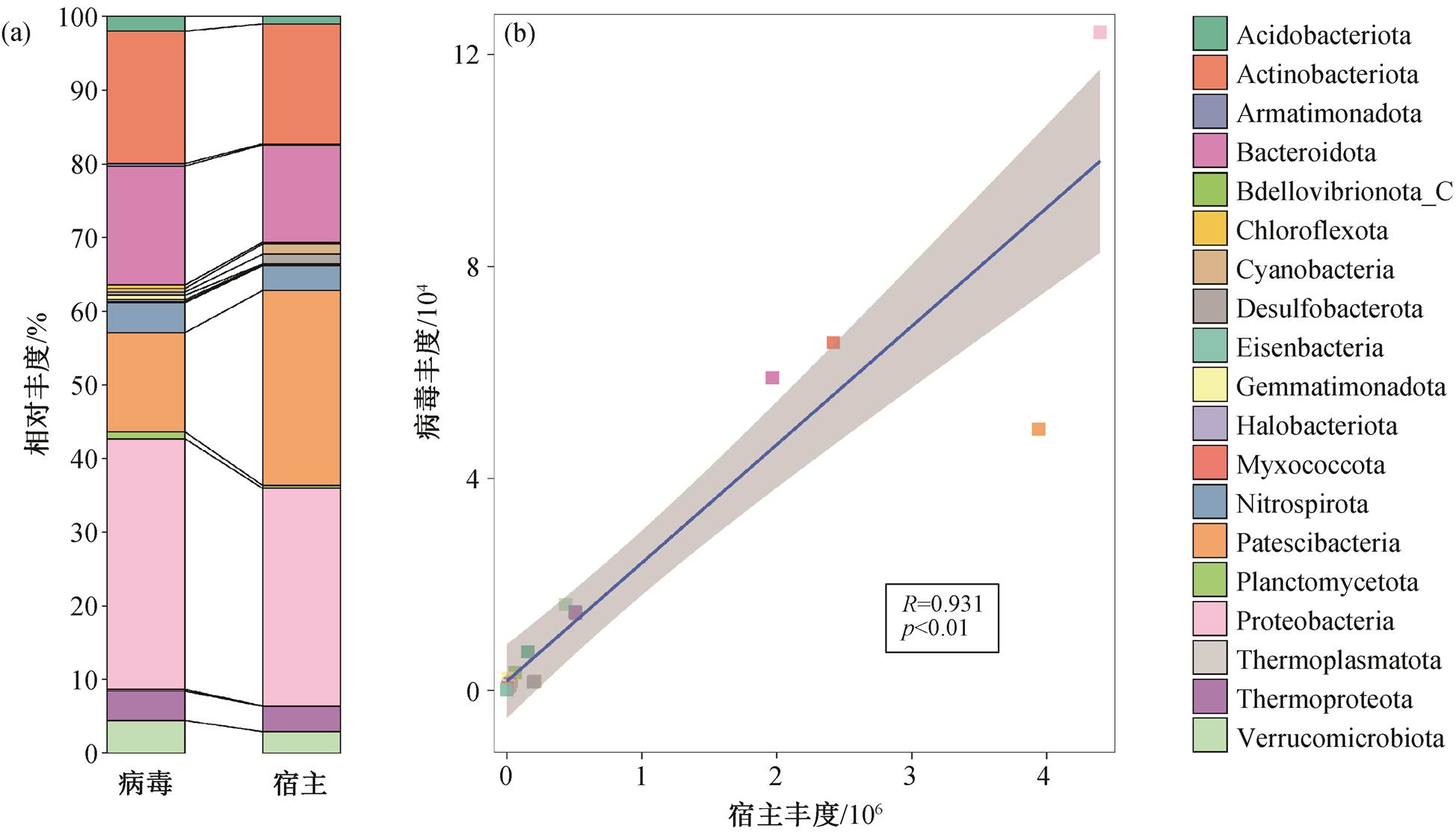
图7 病毒和宿主以宿主门分类计算的相对丰度对应占比(a)和Pearson相关分析(b)
Fig. 7 Relative abundance proportion (a) and Pearson correlation (b) of viruses and their predicted hosts grouped by the host taxonomy
88.4%的病毒具有单一门水平的宿主, 此外的366 个 vOTUs 则注释到多个门的宿主。具体来看, 总体上得到注释的 vOTUs 中, 有 9.2%的 vOTUs 具有跨两个门的宿主, 1.9%的 vOTUs 具有跨 3 个门的宿主, 0.4%的 vOTUs 具有跨 4 个门的宿主, 最多的宿主跨越 5 个门。
共计 39 个 vOTUs 存在跨细菌和古细菌界的感染, 占比为 1.2%。如表 1 所示, 这一类 vOTUs 在感染古菌界 Thermoproteota 门的同时, 最常见于感染细菌界的 Bacteroidota 门(20 个 vOTUs)。
对门水平的宿主群落分析季节变化和空间分布差异, 结果如图 6 所示。丰度最高的两个细菌门Proteobacteria 和 Actinobacteriota 在沉积物中并不占据如水体中的绝对优势, 表明在沉积物中宿主的群落组成更为多样化。与水体相比, Nitrospirota 门、Desulfobacterota 门和 Acidobacteriota 门在沉积物中占据更大的比例。此外, 古菌界 Thermoproteota 门的丰度在沉积物中也比水体中高。在水体中比沉积物中占比更大的为 Verrucomicrobiota 门。与秋季水体相比, 在春季水体中 Bacteroidota 门宿主的占比总体上更大, Acidobacteriota 门和 Planctomycetota 门的占比总体上更小。在 3 号样点, 秋季底泥中的Cyanobacteria门丰度占比较高。
为探究病毒与宿主丰度的关联, 对两者的丰度按宿主门分类。如图 7(a)所示, 病毒与对应宿主的各门相对丰度的占比基本上一致, 尤其是非优势门的宿主类别。其中, 感染 Proteobacteria 门的病毒丰度占比高于宿主, 主要是由于跨门感染大多会感染Proteobacteria 门。图 7(b)显示不同门类病毒和宿主的丰度具有显著的 Pearson 相关性(p<0.01), 进一步证明微生物宿主的组分与病毒的组分在门水平上相当, 与深海沉积物样品的相关研究结论[33]一致。
本文基于 2014 年 3 月和 10 月汉江中下游 6 个断面的水体和沉积物监测样品, 研究噬菌体的特征, 得到如下结论。
1)得到 384 个高质量的 vOTUs, 95%以上隶属Caudovirales 目, 丰度排前三的科分别为 Myovi-ridae, Siphoviridae 和 Podoviridae。
2)沉积物介质中病毒群落具有季节稳定性, 而水体中的病毒群落更容易随季节发生变化。同流域的水体和沉积物病毒群落可能具有连通性; 与不同季节相比, 同一季节的水体与沉积物样品之间的聚类更加紧密。
3)宿主来自 2 域(界) 19 门, 丰度最高的宿主是Proteobacteria 门、Actinobacteriota 门和 Bacteroido-ta 门。88%的噬菌体具有单一门水平的宿主, 有 3个 vOTUs 宿主跨越 5 个门。跨界感染在感染古菌界Thermoproteota 门的同时, 最常见于感染细菌界的Bacteroidota门。
4)与水体相比, 沉积物中宿主的群落组成更加多样化, Nitrospirota 门、Desulfobacterota 门和 Aci-dobacteriota 门宿主在沉积物中占据更大的比例。此外, 古菌界 Thermoproteota 门的丰度在沉积物中比水体中高。在水体中比沉积物中占比更大的为Verrucomicrobiota 门。相比秋季水体, 春季水体中Bacteroidota 门宿主的占比总体上更大, Acidobac-teriota 门和 Planctomycetota 门的占比总体上更小。
5)噬菌体与对应宿主的各门相对丰度占比分布基本上一致, 丰度具有显著的 Pearson 相关性, 表明噬菌体的组分与微生物宿主组分在门水平上相 一致。
参考文献
[1]Suttle C A. Viruses in the sea. Nature, 2005, 437: 356–361
[2]Dion M B, Oechslin F, Moineau S. Phage diversity, genomics and phylogeny. Nature Reviews Microbio-logy, 2020, 18(3): 125–138
[3]Breitbart M, Thompson L R, Suttle C A, et al. Ex-ploring the vast diversity of marine viruses. Oceano-graphy, 2007, 20(Spl. Iss. 2): 135–139
[4]Sano E, Carlson S, Wegley L, et al. Movement of viruses between biomes. Applied and Environmental Microbiology, 2004, 70(10): 5842–5846
[5]Deng L, Ignacio-Espinoza J C, Gregory A C, et al. Viral tagging reveals discrete populations in Syne-chococcus viral genome sequence space. Nature, 2014, 513: 242–245
[6]Martinez-Hernandez F, Fornas O, Lluesma Gomez M, et al. Single-virus genomics reveals hidden cosmopo-litan and abundant viruses. Nature Communications, 2017, 8: 15892
[7]Hurwitz B L, Sullivan M B. The Pacific Ocean Virome (POV): a marine viral metagenomic dataset and associated protein clusters for quantitative viral ecology. PLOS ONE, 2013, 8(2): e57355
[8]Duarte C M. Seafaring in the 21st century: the Mala-spina 2010 circumnavigation expedition. Limnology and Oceanography Bulletin, 2015, 24(1): 11–14
[9]Brum J R, Cesar Ignacio-Espinoza J, Roux S, et al. Patterns and ecological drivers of ocean viral com-munities. Science, 2015, 348: 1261498
[10]Roux S, Brum J R, Dutilh B E, et al. Ecogenomics and potential biogeochemical impacts of globally abundant ocean viruses. Nature, 2016, 537: 689–693
[11]Paez-Espino D, Eloe-Fadrosh E A, Pavlopoulos G A, et al. Uncovering Earth’s virome. Nature, 2016, 536: 425–430
[12]李樾, 袁智, 刘丽. 基于病毒宏基因组技术的浮游病毒多样性研究进展. 湖北工程学院学报, 2020, 40(6): 42–48
[13]Ge X, Wu Y, Wang M, et al. Viral metagenomics analysis of planktonic viruses in East Lake, Wuhan, China. Virologica Sinica, 2013, 28(5): 280–290
[14]Cai L, Zhang R, He Y, et al. Metagenomic analysis of virioplankton of the subtropical Jiulong River Estuary, China. Viruses, 2016, 8(2): 35
[15]李樾. 基于宏基因组学分析云南富营养化湖泊浮游病毒多样性[D]. 昆明: 昆明理工大学, 2017
[16]Krueger F. Trim galore. A wrapper tool around Cutadapt and FastQC to consistently apply quality and adapter trimming to FastQ files [EB/OL]. (2012–03) [2021–12–10]. https://github.com/FelixKrueger/T rimGalore
[17]Uritskiy G V, DiRuggiero J, Taylor J. MetaWRAP — a flexible pipeline for genome-resolved metagenomic data analysis. Microbiome, 2018, 6(1): 1–13
[18]Gregory A C, Zayed A A, Conceição-Neto N, et al. Marine DNA viral macro- and microdiversity from pole to pole. Cell, 2019, 177(5): 1109–1123
[19]Nayfach S, Camargo A P, Schulz F, et al. CheckV assesses the quality and completeness of metagenome-assembled viral genomes. Nature Bio-technology, 2021, 39(5): 578–585
[20]Fu L, Niu B, Zhu Z, et al. CD-HIT: accelerated for clustering the next-generation sequencing data. Bioin-formatics, 2012, 28(23): 3150–3152
[21]Hyatt D, Chen G L, LoCascio P F, et al. Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC Bioinformatics, 2010, 11(1): 119
[22]Bin Jang H, Bolduc B, Zablocki O, et al. Taxonomic assignment of uncultivated prokaryotic virus genomes is enabled by gene-sharing networks. Nature Biotech-nology, 2019, 37(6): 632–639
[23]Pruitt K D, Tatusova T, Maglott D R. NCBI reference sequences (RefSeq): a curated non-redundant sequen-ce database of genomes, transcripts and proteins. Nuc-leic Acids Research, 2007, 35(Suppl. 1): D61–D65
[24]Mende D R, Bryant J A, Aylward F O, et al. Envi-ronmental drivers of a microbial genomic transition zone in the ocean’s interior. Nature Microbiology, 2017, 2(10): 1367–1373
[25]Langmead B, Salzberg S L. Fast gapped-read align-ment with Bowtie 2. Nature Methods, 2012, 9(4): 357–359
[26]Cock P J A, Bonfield J, Chevreux B, et al. SAM/BAM format v1.5 extensions for de novo assemblies. Bio-Rxiv, 2015: 020024
[27]Pons J C, Paez-Espino D, Riera G, et al. VPF-Class: taxonomic assignment and host prediction of uncul-tivated viruses based on viral protein families. Bio-informatics, 2021, 37(13): 1805–1812
[28]Parks D H, Chuvochina M, Waite D W, et al. A standardized bacterial taxonomy based on genome phylogeny substantially revises the tree of life. Nature Biotechnology, 2018, 36(10): 996–1004
[29]Altschul S F, Gish W, Miller W, et al. Basic local alignment search tool. Journal of Molecular Biology, 1990, 215(3): 403–410
[30]Shannon P, Markiel A, Ozier O, et al. Cytoscape: a software Environment for integrated models of bio-molecular interaction networks. Genome Research, 2003, 13(11): 2498–2504
[31]Hanauer D I, Graham M J, Betancur L, et al. An inclusive Research Education Community (iREC): impact of the SEA-PHAGES program on research outcomes and student learning. PNAS, 2017, 114(51): 13531–13536
[32]张奇亚, 桂建芳. 水产动物的病毒基因组及其病毒与宿主的相互作用. 中国科学: 生命科学, 2014, 44 (12): 1236–1252
[33]Li Z, Pan D, Wei G, et al. Deep sea sediments associated with cold seeps are a subsurface reservoir of viral diversity. The ISME Journal, 2021, 15(8): 2366–2378
Characteristics of Phages in Water and Sediments of Hanjiang River
Abstract Based on the water and sediment monitoring samples of 6 sections in the middle and lower reaches of the Hanjiang River in March and October 2014, 384 high-quality vOTUs (viral Operational Taxonomic Units) were obtained. The research showed that more than 95% of the vOTUs belonged to Caudovirales, and the top three abundance families were Myoviridae, Siphoviridae and Podoviridae. The analysis of PCoA (principal coordinates analysis) and ANOSIM(analysis of similarities) showed that the phage community structure in sediments was relatively stable, while the phage community in water was easy to change with seasons, and the phage community in water and sediments of the same basin might be connected. Hosts came from 19 phyla of 2 domains (kingdoms), and the most abundant host was Proteobacteria. 88% of the vOTUs had a single host of phylum level, while three vOTUs exhibited a broad host range across five phyla. Phages infecting hosts across domain (kingdoms) would infect Thermoproteota, and most commonly infected Bacteroidota. Compared with water, the community composition of phage hosts in sediments was more diverse. Pearson correlation analysis showed that the composition of pahges agreed with that of their microbial hosts at the phylum level.
Key words Hanjiang River; phage; host; water; sediments