doi: 10.13209/j.0479-8023.2022.087
北京大学学报(自然科学版) 第58卷 第5期 2022年9月
Acta Scientiarum Naturalium Universitatis Pekinensis, Vol. 58, No. 5 (Sept. 2022)
收稿日期: 2021-10-12;
修回日期: 2021-12-12
摘要 基于密度泛函理论中的第一性原理计算方式, 开展 KDP(100)面表面吸附水分子的性质研究。结合Bader 电荷、电子密度、差分电子密度和电子局域函数等参数进行电子密度拓扑分析, 结果显示水分子在KDP(100)表面的最佳吸附位点为氢钾桥位, 吸附能为–0.809eV, 表明 KDP(100)表面可以自发地吸附水分子; 水分子中的氧原子通过吸附, 与 KDP(100)表面磷酸根基团上的氢原子形成含共价效应的强氢键 O—HOw, 拟合键能为–18.88 kcal/mol。
关键词 KDP 晶体; 密度泛函理论; 第一性原理分子动力学; 电子密度拓扑分析; 吸附
当今世界对能源的需求与日俱增, 而目前大规模使用的化石燃料是不可再生能源, 这就驱使着人们寻找更加高效的新能源。其中, 可控核聚变因燃料储量巨大以及产物无污染等性质而受到广泛关注。惯性约束核聚变(ICF)是实现可控核聚变的途径之一。该方法通过使用大功率激光器, 从各个方向对燃料靶丸进行压缩加热, 从而达到核聚变的点火条件。KDP 晶体因其优秀的非线性光学性质以及较高的激光损伤阈值, 广泛用作大功率激光器中的倍频光学器件, 其晶体生长及其光学性质一直是研究热点。Bacon 等[1]基于中子衍射的方法, 研究KDP 晶体的内部结构。Yoreo 等[2]开发出一种高效生长 KDP 晶体的制备方法, 并定量地研究 KDP 晶体缺陷与光学性能之间的关系。Fu等[3]对 KDP 培养液中各种阴离子添加剂(如 PO43–和 H2PO4–等)与KDP 晶体生长的关系进行研究, 结果表明所研究的各种阴离子均对 KDP 锥面体的生长产生抑制效果。Fujioka 等[4]使用点状籽晶快速生长法, 生长出64mm×63mm×43mm 的 KDP 晶体, 并对其光学性质进行检测, 结果表明, 相对于传统的制备方法, 该KDP 晶体的棱柱(100)面吸附了更多的金属阳离子, 如 Fe3+, Al3+和 Na+等。
KDP 在空气中具有易潮解的特性, 使得相关光学器件在一段时间后出现光学性质改变的现象, 导致激光损伤阈值大幅下降, 影响大功率激光器的正常运行, 甚至造成损坏[5]。所以, 研究水分子与 KDP表面的潮解机理与性质具有重要意义。Yin 等[6]的研究结果表明, 潮解作用机理为空气中的微液滴发生微凝结效应, 吸附于固体表面, 造成局部再溶解与再蒸发, 导致晶体腐蚀。他们还提出, 降低晶体与外界流体的温度差可以有效减慢上述潮解循环, 从而抑制潮解作用的发生。张秀丽[7]利用潮解作用中的溶解性质, 将潮解作用应用于 KDP 晶体的抛光, 并通过实验对比配制出合适的抛光液, 得到较好的抛光效果。Liu 等[8]提出将两相空气–水流体(TAWF)作为催化剂的抛光技术, 实现 KDP 晶体的可控潮解, 对 KDP进行高效的潮解抛光处理。
密度泛函理论(DFT)因其计算精确以及不依赖于经验公式的特性, 广泛应用于对 KDP 晶体的各类性质研究中。Mylvaganam 等[9]基于密度泛函理论计算, 探究不同晶轴上应力对 KDP 晶体结构与相变的影响。Zhou 等[10]使用密度泛函理论计算与实验结合的方法, 研究 KDP 和 ADP 晶体的结构以及表面声波特性等相关物理性质。Koval 等[11]将该方法运用于研究 KDP 晶体的铁电性及其氘化后产生的相应同位素效应, 同时表明其原理为氘化后 O—H—O 桥中心质子概率密度减小。
在计算晶体表面吸附小分子的问题上, 研究人员借助 DFT 也做出相当多的成果。Bromfield 等[12]基于 DFT 计算, 探究 CO 分子在 Fe(100)面上的吸附。Bolton 等[13]分别研究水分子在 Ni(111), Ni(110)和 Ni(100)3 种晶体表面构型上的吸附。Wu 等[14]将该理论用于计算诸如 K+, Na+和 Al3+等各类阳离子在 KDP(100)表面上的吸附性质, 结果表明各类阳离子均不同程度地自发地吸附到晶体表面上。
但是, 对于将上述两者结合的研究(即 KDP 表面吸附水分子的 DFT 计算)较为缺乏。Zhang 等[15]基于第一性原理计算, 通过将水分子置于不同初始位点进行结构弛豫, 并进行对比, 研究水分子在KDP(100)表面和 KDP(101)表面的最佳吸附方式, 发现在(100)表面, 会根据吸附位点的不同, 形成氢键, 或氢键和氢钾键; 对(101)表面, 则会同时形成氢键和氢钾键。但是, 该研究未在计算中加入分子间作用力修正项, 对成键的判断也过于定性化。从总体上看, 对水分子在 KDP 表面吸附的研究仍然较少, 对 KDP 晶面吸附水分子后的相关物理性质计算更是鲜有报道。
Hartman[16]利用周期键链(PBC)理论对 KDP 晶体的理想外形进行理论预测, 表明在实际制备过程中, KDP 晶体表面只会形成棱柱(100)类型面和棱锥(101)类型面两种平坦表面构型。Vries 等[17]通过对 KDP 表面进行 X 射线衍射探测, 在对Hartman 的理论进行验证的同时, 进一步确定两种表面的终止结构: 对于(101)类型面, 其表面最外层以 K+阳离子为终止; 对于(100)类型面, 其唯一的终止界面为中性层, 层级之间依靠氢键 O—HO 相互结合。在上述研究的基础上, 且因自然状态下的 KDP(100)面为在空气中的主要显露面之一, 本文选择棱柱(100)面作为研究对象。
本文使用第一性原理分子动力学模拟的方法, 以更加底层的手段确定 KDP(100)表面上的水分子最佳吸附位点, 并通过计算吸附能, 得出水分子与表面相互作用为一种物理吸附, 意味着可以较容易地通过各种后期抛光手段解决潮解问题。此外, 通过计算电子密度分布、差分电子密度、ELF 电子函数、Bader 电荷布居、态密度 以及电子密度拓扑, 对水分子在 KDP (100)表面上的成键机理进行探究, 以期对 KDP 晶体潮解过程的研究乃至大型激光器的维护提供理论依据。
本文的计算均使用基于密度泛函理论(DFT)[18]的Vienna Ab-initio Simulation Package (VASP)[19]程序包 5.4.1 版本, 通过求解 Kohn-Sham (K-S)[20]方程的方式完成。在求解 K-S 方程的过程中, VASP 程序包采用基于平面波基组展开的投影缀加平面波 (projector augmented wave, PAW) [21]方法, 计算得到全电子波函数。本文的计算体系中, H 的价电子取 1s1, O 的价电子取 2s22p4, P 的价电子取 3s23p3, K的价电子取 3s23p64s1。在交换关联泛函方面, 本文选择选择广义梯度近似(generalized gradient appr-oximation, GGA)方法下的Perdew-Burke-Ernzerhof (PBE) [22]泛函来描述交换关联作用。经过能量收敛测试, 计算的平面波截止能设置为 450eV, 布里渊区的 k 点网格划分采用Monkhorst-Pack (MP) [23]方法。在进行 KDP 晶体的优化计算、电子相关计算以及态密度计算时, 使用带 Blöchl 修正的四面体方法来确定每个电子波函数的部分占有率; 在进行其他计算时, 使用 Gaussian smearing 法确定每个电子波函数的部分占有率, 展宽设置为 0.05eV。
KDP 晶体在常温常压下呈四方晶系, 空间群为I![]() 2d, 点群为 D2d。单胞晶格常数 a = b = 7.453Å, c = 6.972Å, ɑ = β = γ = 90°[24]。在目前的制备工艺下, KDP晶体的理想表面由 4 个四棱柱面和 8 个四棱锥面构成, 其中棱柱面为(100)面, 棱锥面为(101)面[16]。本文以棱柱面(100)面为例, 探究其对水分子的吸附性质和机理。
2d, 点群为 D2d。单胞晶格常数 a = b = 7.453Å, c = 6.972Å, ɑ = β = γ = 90°[24]。在目前的制备工艺下, KDP晶体的理想表面由 4 个四棱柱面和 8 个四棱锥面构成, 其中棱柱面为(100)面, 棱锥面为(101)面[16]。本文以棱柱面(100)面为例, 探究其对水分子的吸附性质和机理。
为了构建表面模型, 我们首先按照相关 KDP 晶体参数(表 1)构建单胞计算模型。然后对构建的模型进行结构优化。在对 KDP 晶体的结构优化时, 对k 点采用 5×5×5 网格, 收敛判据为原子间受力小于0.01eV/Å, 体系总能量变化小于 1.0×10–5eV/atom。本文充分考虑了范德华作用对计算的影响。目前主流的对 DFT 中范德华作用的修正方案有两种: 一种是在交换关联能时, 加入一项长程关联能 vdW-DF和 vdW-DF2[25]; 另一种是在总能中加入一个经验的原子对间的两两色散作用项 DFT-D3[26]。DFT-D3 方法的能量修正项又可分为基于 Becke-Johnson (BJ)阻尼函数[27]的形式和基于零阻尼的形式。本文以优化 KDP 晶体结构为例, 系统地对比在该体系下使用 vdW-DF2 方法、基于 BJ 阻尼函数的 DFT-D3 修正方法、基于零阻尼的 DFT-D3 修正方法以及不添加范德华修正项这 4 种方法的计算结果(表 1)。
由表 1 可知, 3 种范德华修正方法和不做修正的计算结果与实验值的误差均在 1%以内, 都在可信范围内, 说明无论哪种方法都可以得到可靠的结果。进一步分析可以发现, 基于零阻尼的 DFT-D3方法与参考值的误差最小。同时, 考虑到 DFT-D3方法所需的额外计算量极少, 本文所有计算均使用基于零阻尼的 DFT-D3 方法进行范德华修正。
在充分优化的 KDP 晶体模型基础上, 我们构建KDP(100)面物理模型, 方法如下: 将初始单胞沿 Z轴拓展 3 倍, 然后沿(100)面对拓展后的超胞进行切割。因切割深度(或终止表面类型)不同, 并考虑到对称性, 共有 6 种不同的构型(1~6 号构型), 如图 1 所示。但是, 考虑到 KDP 晶体内的磷酸基团各原子间为共价键连接, 键能较大, 在该基团内部切割构型不易稳定存在, 又根据 Vries 等[17]的 X 射线衍射测定结果, 实际上制备出的 KDP 晶体(100)表面只会存在 4 号构型这一种类型。为验证这一点, 首先构建上述 6 种切割构型, 各种构型的表面结构均由 3 层 XY 面层构成, 切割表面与 Z 轴垂直。为正确地模拟表面结构, 防止 Z 轴方向相邻的周期性结构产生彼此间的相互作用, 在每种构型表面均构建厚度为 30Å 的真空层。在该超晶胞结构下, 对 k 点采用 5×5×1 的网格, 固定除表面 3 层原子外的原子, 对每种构型进行充分的优化。经测试, 该真空层厚度下各种构型的体系能量收敛良好, 说明真空层厚度的取值可靠。各种构型优化后的总能量如表 2 所示, 可知 4 号构型的总能最低, 即为最稳定结构, 与上述理论预测相符, 故本文的计算都基于 4 号构型进行。
我们对水分子结构也进行优化。将一个水分子置于 10Å×10Å×10Å 的真空盒子中, 使用与优化KDP 晶体相同的能量收敛判据等参数对该水分子进行优化。优化后, 得到的水分子键角为 104.44°, 键长为 0.972Å, 与实验值 104.48°和 0.958Å[28]对比, 键角相差 0.04%, 键长相差 1.46%, 均基本上吻合, 进一步证明计算参数的可靠性。
为探究水分子在各种构型下 KDP(100)面的最佳吸附位点, 本文采用第一性原理分子动力学模拟与结构优化相结合的方式观察末态, 并对水分子的吸附位点进行结构优化, 以此作为整个吸附过程的最佳吸附位点。首先将水分子平行置于优化好的KDP (100)表面上方 5Å 处, 并固定除水分子和(100)表面 3 层原子外的其他原子, 以此作为初态进行分子动力学计算, 如图 2(a)所示。为加快吸附进程, 对水分子施加一个方向垂直于(100)表面的极小的初始速度, 其初动能与 300K 下水分子的平均热动能相当。模拟系统选用正则(NVT)系综, 温度设定为 300 K, 并使用 Nose-Hoover 热浴方法控制系统的温度近似恒定。这里对 k 点采用 4×4×1 的网格, 收敛判据为原子间受力小于 0.01eV/Å, 体系总能量变化小于 1.0×10–4eV/atom。模拟计算共持续 5000 步, 步长为 1fs, 共 5000fs, 第 5000 步时的构型如图 2 (b)所示。同时, 如图 2(c)所示, 通过系统总能变化趋势进行分析, 系统在第 1000fs 后能量的下降趋于稳定, 证明模拟所选取的时间范围是可靠的。
表1 3种范德华修正与实验值的偏差对比(Å)
Table 1 Comparison of deviations between 3 kinds of van der Waals corrections and experimental values(Å)
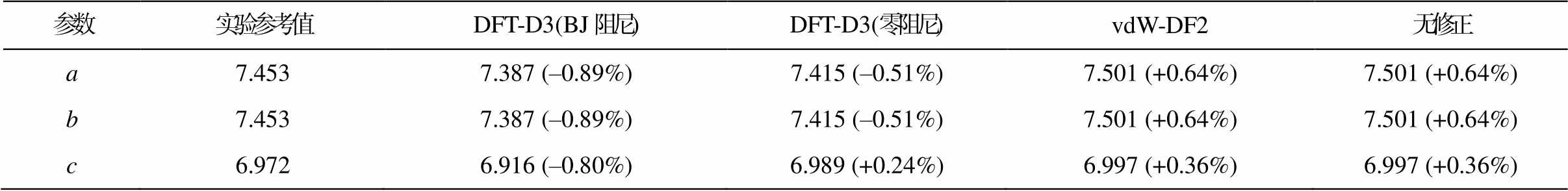
参数实验参考值DFT-D3(BJ阻尼)DFT-D3(零阻尼)vdW-DF2无修正 a7.4537.387 (–0.89%)7.415 (–0.51%)7.501 (+0.64%)7.501 (+0.64%) b7.4537.387 (–0.89%)7.415 (–0.51%)7.501 (+0.64%)7.501 (+0.64%) c6.9726.916 (–0.80%)6.989 (+0.24%)6.997 (+0.36%)6.997 (+0.36%)
说明: 括号中数据为与实验值的误差。
表2 不同终止表面下体系结构优化后的总能
Table 2 Total energy after system structure optimization with different termination surfaces

构型编号123456 总能量/eV–556.867–567.942–565.102–576.278–571.412–564.495
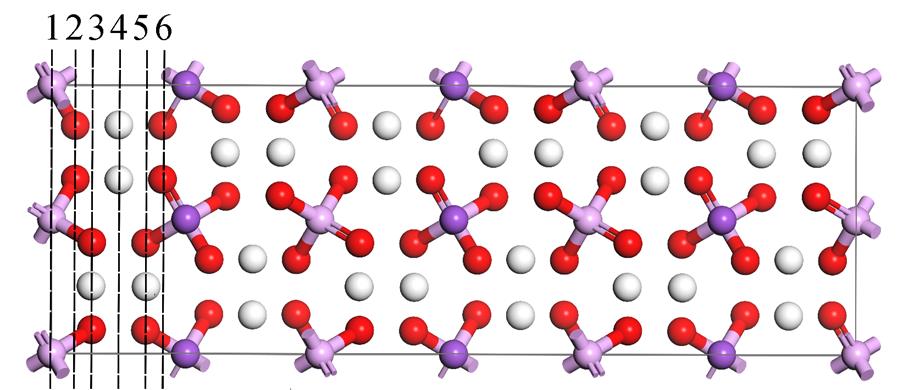
图1 KDP单晶拓展超胞后6种不同切割深度示意图
Fig. 1 Schematic diagram of 6 different cutting depths after KDP expanding

图2 初态构型(a)、第 5000 步时构型(b)和分子动力学过程中系统总能变化(c)
Fig. 2 Configuration at step 0 (a), configuration at step 5000 (b) and curve of total energy of the system during the molecular dynamics process (c)
进一步地, 我们对分子动力学计算得到的末态进行结构优化, 并计算该吸附构型下的吸附能, 计算参数与优化 KDP(100)面时相同, 末态构型见图2。吸附能计算公式如下:
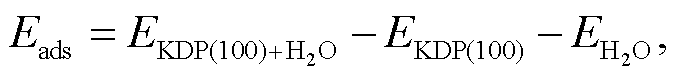 (1)
(1)
其中, EKDP(100)+H2O 代表吸附水分子后的KDP(100)表面的体系总能量; EKDP(100)代表未吸附水分子时的清洁的 KDP(100)表面的总能量; EH2O 代表单个水分子的能量; Eads 代表该模型下, 经过分子动力学计算和结构弛豫, KDP(100)表面吸附 H2O 分子后的体系总能量, 该能量可以作为表面吸附能力强弱的判断依据, Eads 越低, 代表吸附后的体系越稳定, 即 H2O 越容易吸附在KDP(100)表面上。
根据计算结果, EKDP(100)+H2O 为–591.309eV, EH2O为–14.222eV, 由表 2 可得 EKDP(100)为–576.278eV, 则由式(1)可得该构型下对水分子吸附能为–0.809 eV。说明在常温环境下, 水分子会较容易地自发性吸附在 KDP(100)表面, 导致潮解现象的发生。
图 3(a), (c)和(e)表示经过 5000 步分子动力学模拟和结构优化后, 体系末态构型的三视图; 图 3(b), (d)和(f)表示在无水分子加入时, 经过结构优化的体系初态三视图。对图 3 中吸附前后的表面构型进一步分析可知, 在优化后的稳定构型下, 水分子中的氧原子 Ow 与 KDP(100)表面磷酸根基团上的最优吸附位点为 H1—K1 桥位, 其中与氢原子 H1 距离为1.578Å, 与钾原子 K1 之间的距离为 3.013Å。KDP (100)表面磷酸根基团上的氧原子 O1 与氢原子 H1 所成键 O1—H1, 与 Ow 所成的角度为 162.899°, 接近180°。同时, 水分子键角由初态的 104.44°增大为106.377°, Ow—Hw1 键长由初态的 0.972Å 增大为0.987Å, Ow—Hw2 由初态的 0.972Å 增大为 0.995Å。KDP (100)表面的磷酸根基团中距水分子近侧的 O1—H1键长由 0.996Å 增长为 1.030Å, 并向水分子中的氧原子 Ow 方向移动, 表现出明显的被水分子吸引的特征。晶体表面磷酸根基团的两根磷氧共价键 P1—O1 和 P2—O2 键长分别由初态的 1.625Å 变为 1.590Å 和 1.628Å。据上述几何构型分析, 推测在该两原子之间可能形成 O1—H1Ow 形式的氢键, 而该氢键的形成导致水分子中两条氢氧共价键和KDP(100)表面磷酸根基团的氢氧共价键 O1—H1 遭到明显的削弱。
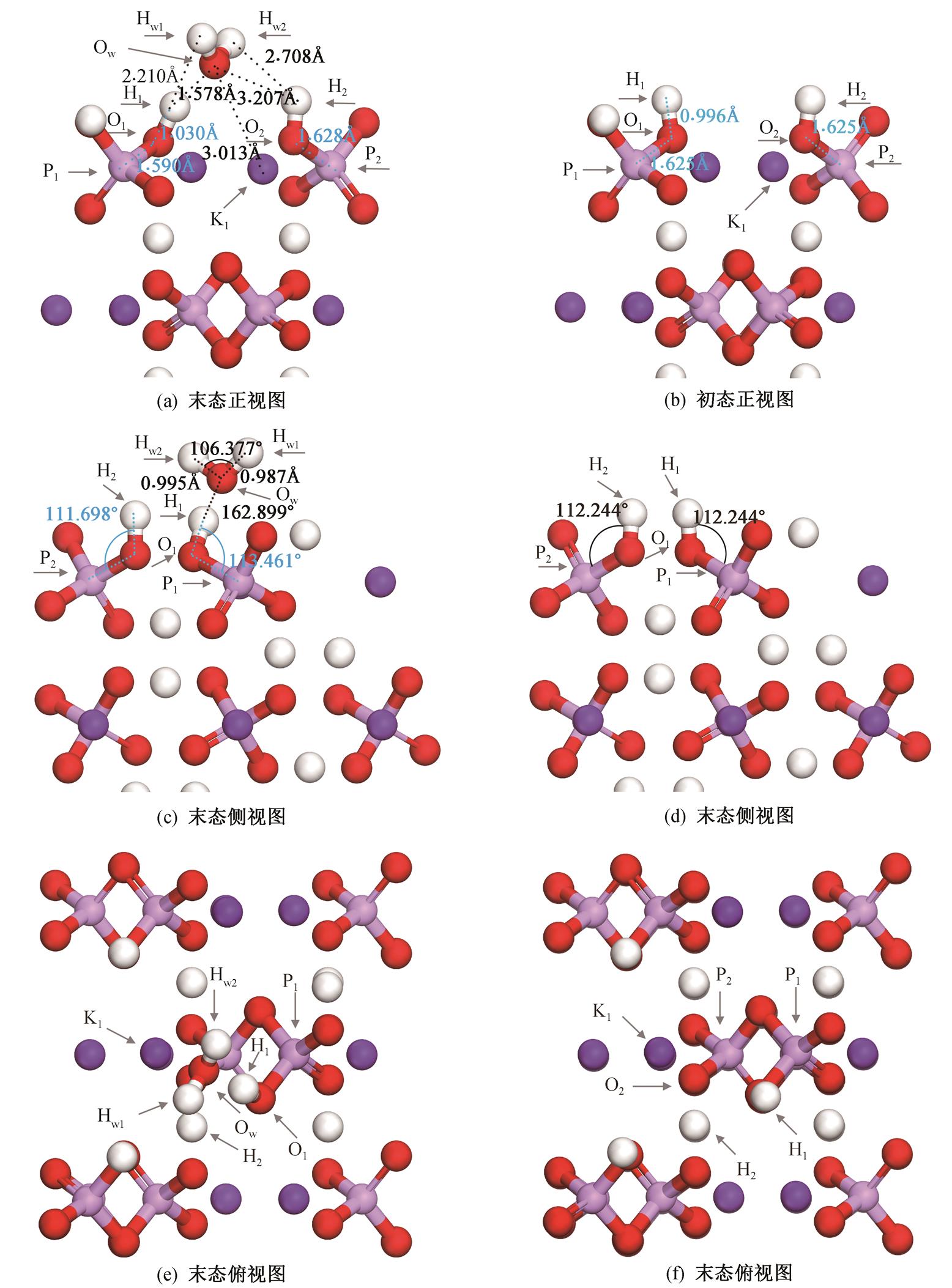
图3 末态分子构型三视图
Fig. 3 Three views of the final molecular configuration
我们同时对吸附后体系总态密度和分波态密度进行计算, k 点网格取为 9×9×1。图 4 为系统的总态密度以及 Ow 原子、H1 原子和 K1 原子的相关分波态密度图。
从图 4 可以看出, KDP 表面磷酸根基团中的氢原子 H1-1s 轨道与水分子中的氧原子 Ow-2s 轨道在–21.2eV 处存在共振峰, 但由于该处距费米能级较远且共振程度非常微小, 可视为对成键没有贡献。H1-1s 轨道与 Ow-2p 轨道在–7.9 eV 和–5.9 eV 处均形成共振峰, 且距离费米能级相对较近, 意味着两原子轨道间可能存在某种程度的杂化, 产生共价作用。磷酸根基团中的钾原子 K1-2p 轨道与 Ow-2s 和Ow-2p 轨道在–5.5eV 至–2.0eV 范围内存在重叠区域, 但由于重叠程度较小, 不存在明显的共振峰, 故共价作用不明显, 可能存在其他相互作用。
由于构型分析与分波态密度分析较为定性化, 为进一步探究水分子与 KDP(100)表面的相互作用与成键情况, 本文同时对体系初末态的电荷进行分析。首先计算该体系末态的差分电子密度图,计算公式如下:
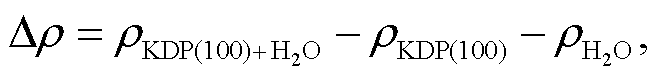 (2)
(2)
其中, 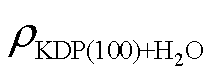 代表 KDP(100)表面吸附 H2O 分子后体系各处的电子密度分布;
代表 KDP(100)表面吸附 H2O 分子后体系各处的电子密度分布; 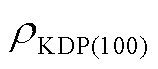 代表当末态构型去掉水分子且其余原子位置不变时, 清洁的KDP(100)体系各处的电子密度分布;
代表当末态构型去掉水分子且其余原子位置不变时, 清洁的KDP(100)体系各处的电子密度分布;  代表末态构型去掉 KDP(100)表面所有原子且水分子位置不变时, 该水分子在体系各处的电子密度分布;
代表末态构型去掉 KDP(100)表面所有原子且水分子位置不变时, 该水分子在体系各处的电子密度分布;  代表该构型末态的差分电子密度, 通过观察, 可以对原子间的相互作用以及成键情况有较为定性和直观的展示。差分电子密度的计算结果如图 5(a)~ (c)所示。在三维差分电子密度图中, 黄色部分代表该区域电子密度增加, 蓝色部分代表该区域电子密度减小, 等势面设置为 0.002 e/bohr3。
代表该构型末态的差分电子密度, 通过观察, 可以对原子间的相互作用以及成键情况有较为定性和直观的展示。差分电子密度的计算结果如图 5(a)~ (c)所示。在三维差分电子密度图中, 黄色部分代表该区域电子密度增加, 蓝色部分代表该区域电子密度减小, 等势面设置为 0.002 e/bohr3。
AIM 理论[29]是一种基于电子密度拓扑性质分析化学键的方法。该理论中, 两原子间的键临界点(BCP)指电子密度标量场梯度为 0, 且相应 Hessian 矩阵的 3 个本征值中有两个为负, 一个为正的点。该点的电子密度 ρ(BCP)、电子密度的拉普拉斯值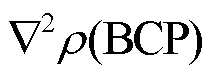 和能量密度 H(BCP)等参数对分析成键有重要意义。对于计算得到的体系电子分布数据, 从两原子连线中点出发, 通过牛顿法多次迭代即可确定两成键原子相应的 BCP 点位置。为了更加直观地展示相应原子间的电子密度及其变化情况, 本文绘制以图 5 中 slice 面为切面的二维差分电子密度图、二维电子局域函数图(ELF)[30]及其二维电子密度图, 如图 5(d)~(f)所示。在划定 slice 面时, 应该使 Ow 原子与 H1 原子之间的键临界点落在 slice 平面内。
和能量密度 H(BCP)等参数对分析成键有重要意义。对于计算得到的体系电子分布数据, 从两原子连线中点出发, 通过牛顿法多次迭代即可确定两成键原子相应的 BCP 点位置。为了更加直观地展示相应原子间的电子密度及其变化情况, 本文绘制以图 5 中 slice 面为切面的二维差分电子密度图、二维电子局域函数图(ELF)[30]及其二维电子密度图, 如图 5(d)~(f)所示。在划定 slice 面时, 应该使 Ow 原子与 H1 原子之间的键临界点落在 slice 平面内。
如图 5 所示, slice 面经过水分子中的氧原子 Ow、KDP(100)表面磷酸根基团中的氢原子 H1 和氧原子O1 以及表面的钾原子 K1。对于二维差分电子密度图和二维电子密度图, 刻度单位均为 e/bohr3, 相邻等高线之间的差间隔分别为 0.001e/bohr3 和 0.001 e/bohr3; 红色部分表示该区域电子密度增大、电子密度大; 蓝色部分表示该区域电子密度减小、电子密度小。对于二维电子局域函数图, 相邻等高线之间的差间隔为 0.1, 蓝色部分表示 ELF 函数值小, 红色部分表示ELF函数值大。
由图 5 可以看出, 由于吸附过程, 水分子与KDP(100)表面相邻的两个磷酸根基团之间都产生较明显的电子密度变化。其中, 与磷酸根基团中的氢原子 H1 附近电子密度减小, 水分子中的氧原子 Ow 以及 Ow 与 H1 之间的区域电子密度则显著增加。为进一步探究不同原子的电子得失情况, 我们对吸附前后的体系分别进行 Bader 电荷布居分析, 该方法以电子密度的零通量面为分界面来区分不同的原子, 结果如表 3 所示。
由表 3 可知, 吸附前后水分子整体上得到 0.2610个电子, 其中氧原子 Ow 得到 0.1546 个电子; 氢原子 H1 失去 0.2727 个电子。这证明在整个吸附过程中, 在两原子间乃至水分子与 KDP(100)晶体表面间存在电子转移。其中氧原子电负性较强, 在吸附过程中得到电子; 氢原子 H1 失去电子, 这部分电子可以解释为在两原子之间的区域, 与氧原子价层电子中的孤对电子在一定程度上形成共用电子对, 导致有微弱的共价作用产生。同时, 钾原子 K1 也失去 0.0421 个电子, 故推测有钾原子也因吸附过程产生某种相互作用。这些电荷转移导致 KDP 表面磷酸基团中电子的分布情况也产生细微的变化。
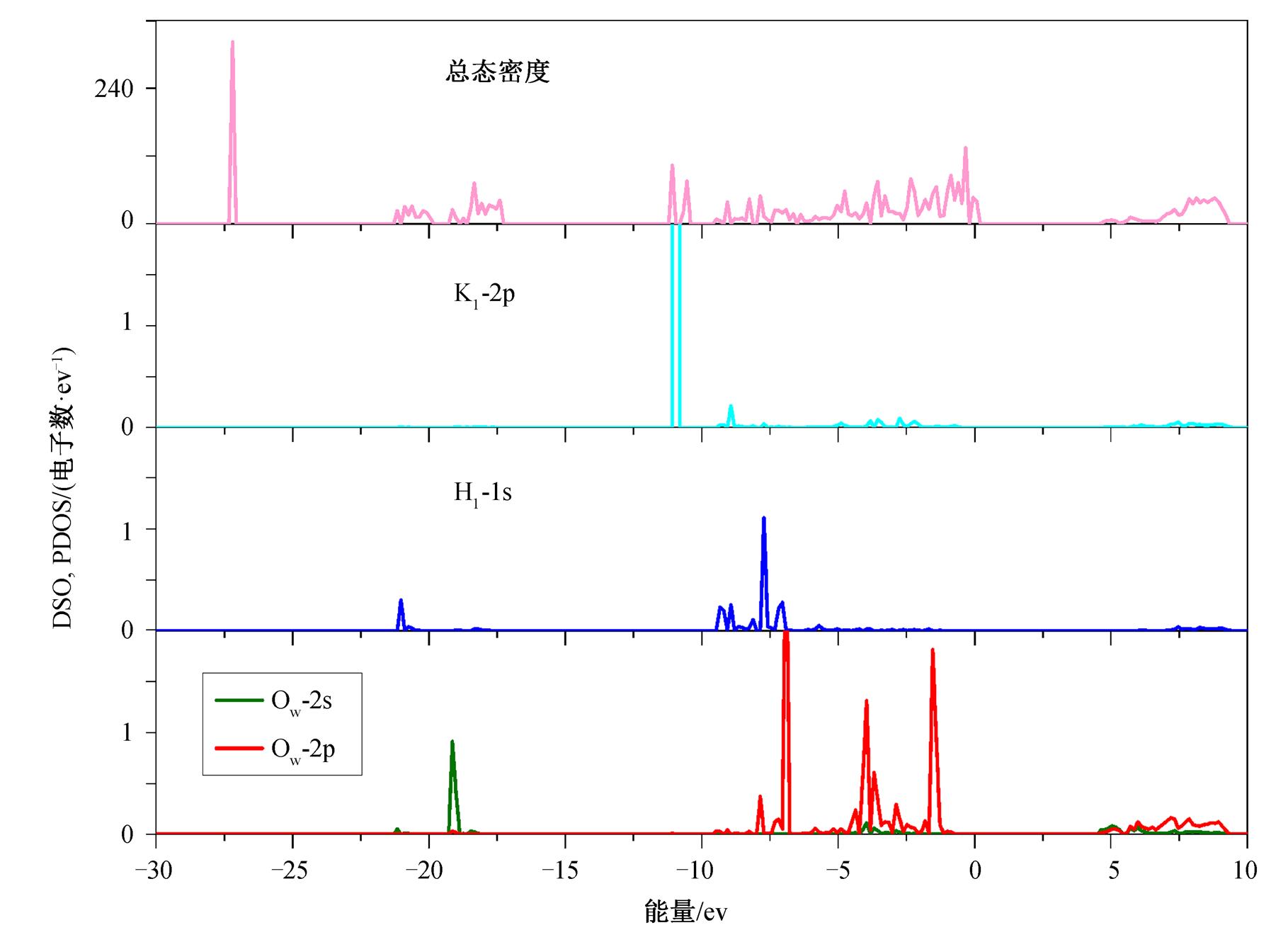
图4 末态分子构型总态密度和分波态密度
Fig. 4 TDOS and PDOS of final state molecular configuration
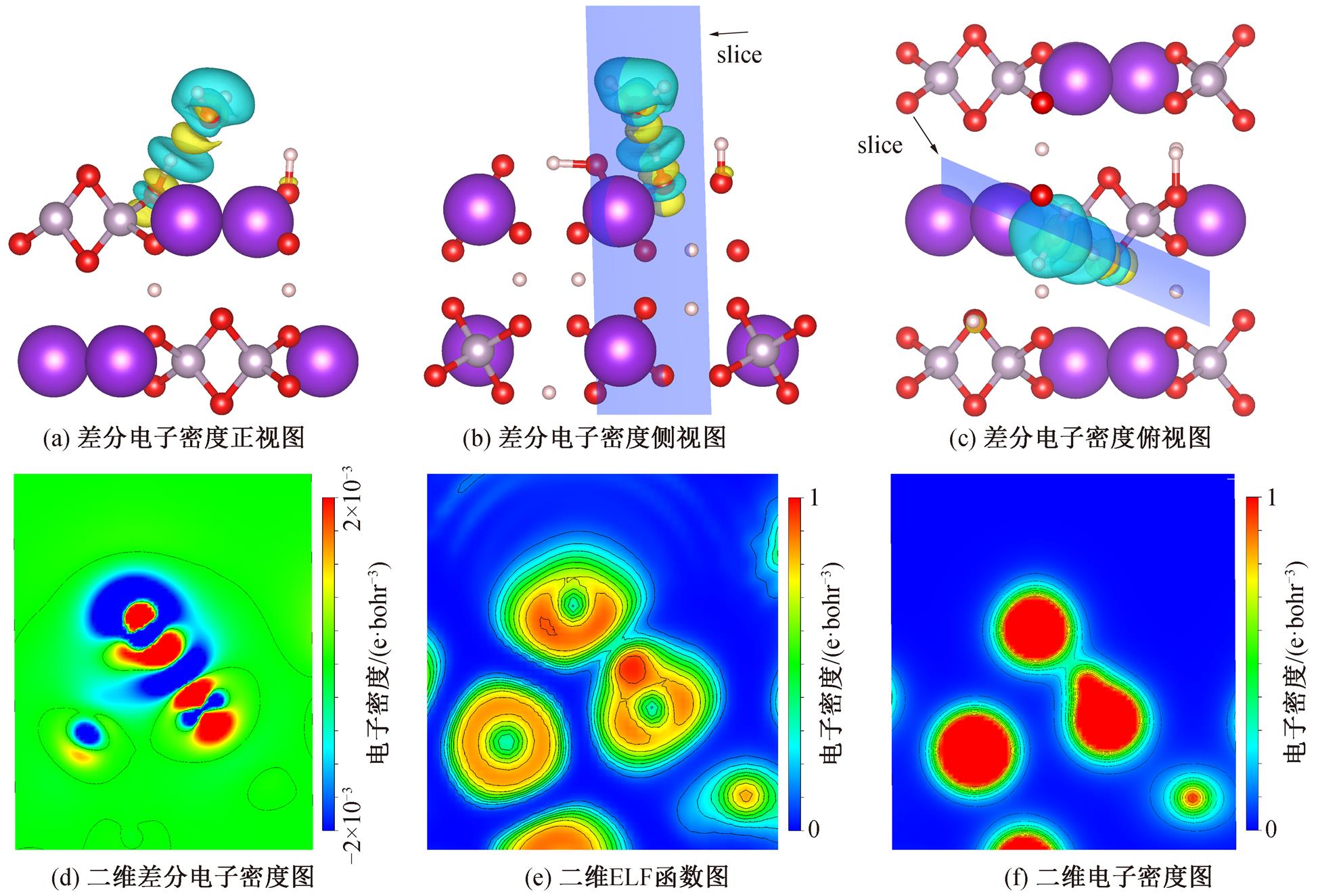
图5 末态电荷分析图
Fig. 5 Charge analysis of final state
表3 不同相关原子的Bader电荷布居分析
Table 3 Bader charge population analysis of different related atoms

吸附情况OwHw1Hw2H1O1K1 吸附前–1.18300.59800.58500.3931–1.43550.8302 吸附后–1.33760.51110.56550.6658–1.46210.8723 净电荷变化–0.1546–0.0869–0.01950.2727–0.02660.0421
说明: 电荷值为负表示该原子得到电子, 电荷值为正代表该原子失去电子。
两原子 BCP 处的电子密度拉普拉斯值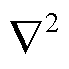 ρ(r)BCP和能量密度 He(r)BCP 等参数可以作为判断成键特征的指标。在大多数情况下, ρ(r)BCP>0 代表该处电子密度发散, 两原子间存在闭壳层相互作用, 如氢键和离子键等; ρ(r)BCP<0 代表该处电子密度聚集, 两原子间存在共价键。在计算过程中, H1Ow键 BCP 点处的ρ(r)BCP 为 0.121 e/bohr5, 为正值, 说明 H1 和 Ow 原子之间不存在纯粹的共价键。He (r)BCP为–0.017hartree/bohr3, 为负值, 代表该处电子势能较低, 电子有向该处聚集的趋势, 即两原子间又存在一定的共价作用, 而非纯粹的闭壳层相互作用。Espinosa 等[31]认为, 若ρ(r)BCP>0 且 He(r)BCP<0,则形成过渡闭壳层相互作用, 即成键性质介于闭壳层作用与共价键之间。他们同时提出键度 BD (bond degree)的概念, 即 BD=He(r)BCP/ρ(r)BCP 表示在键临界点处单位电子的能量密度。BD 可作为共价程度的判据: BD<0 时, 绝对值越大则共价作用越强; BD>0 时, 绝对值越大则共价作用越弱。该体系下的上述参数如表 4 所示。
ρ(r)BCP和能量密度 He(r)BCP 等参数可以作为判断成键特征的指标。在大多数情况下, ρ(r)BCP>0 代表该处电子密度发散, 两原子间存在闭壳层相互作用, 如氢键和离子键等; ρ(r)BCP<0 代表该处电子密度聚集, 两原子间存在共价键。在计算过程中, H1Ow键 BCP 点处的ρ(r)BCP 为 0.121 e/bohr5, 为正值, 说明 H1 和 Ow 原子之间不存在纯粹的共价键。He (r)BCP为–0.017hartree/bohr3, 为负值, 代表该处电子势能较低, 电子有向该处聚集的趋势, 即两原子间又存在一定的共价作用, 而非纯粹的闭壳层相互作用。Espinosa 等[31]认为, 若ρ(r)BCP>0 且 He(r)BCP<0,则形成过渡闭壳层相互作用, 即成键性质介于闭壳层作用与共价键之间。他们同时提出键度 BD (bond degree)的概念, 即 BD=He(r)BCP/ρ(r)BCP 表示在键临界点处单位电子的能量密度。BD 可作为共价程度的判据: BD<0 时, 绝对值越大则共价作用越强; BD>0 时, 绝对值越大则共价作用越弱。该体系下的上述参数如表 4 所示。
电子局域函数图中, Ow 和 H1 原子间的 BCP 处ELF 值为 0.332, 即电子在该点处及其附近区域有一定程度的定域化, 且差分电子密度图中 Ow 与 H1原子之间有电子密度增加的区域, 存在电子云重叠现象, 进一步说明该氢键具有一定程度的弱共价作用。同时, ELF 值小于 0.5也表明该处定域化程度不足以使两原子之间形成典型的共价键, 即该处仍是一种短强氢键。根据以上分析可知, H1Ow 氢键中相互作用的贡献者既有经典氢键间的静电作用, 也有供受体间电子转移产生的部分共价作用。
若H原子和 Y 原子之间形成氢键, 会存在如下性质[32]。
1)参与形成氢键作用力的来源包括静电作用力、由供体和受体之间的电荷转移导致 H 和 Y 之间产生的部分共价键形成的力以及由色散作用引起 的力。
2)X—HY 的角度通常接近 180°, 且 H 原子与Y原子距离越近, 氢键作用越强。
3)X—H 键的长度通常随氢键的形成而增加。X—H键长增加得越多, HY 氢键就越牢固。
4)X 与氢原子之间形成的 X—H 键是极性键, HY 的强度随X电负性增加而增加。
根据构型分析以及电荷分析, 该模型中的O1—H1Ow 键性质与上述氢键性质均较为吻合, 可以判定, 水分子 Ow 与 KDP(100)表面由磷酸根基团提供的顶位氢原子 H1 在吸附过程中确实形成了 O1—H1Ow形式的氢键, 并且该氢键是存在部分共价作用的短强氢键。
对于图 5(e)和(f)继续分析该氢键的 H1 和 Ow 原子, 该处 ρ(BCP) 值为 0.067e/bohr3。可以通过 BCP处的电子密度较为精确地预测中性体系的氢键键能[33], 计算公式为
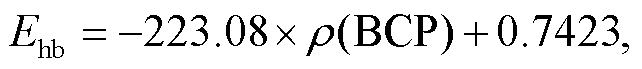 (3)
(3)
其中, Ehb 为氢键键能, 单位为 kcal/mol; 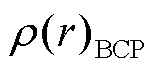 单位为 e/bohr3。将 O1—H1Ow 键的相应电子密度值代入式(3), 可得形成 O1—H1Ow 的氢键键能估计值为–18.88 kcal/mol, 属于强氢键范畴。
单位为 e/bohr3。将 O1—H1Ow 键的相应电子密度值代入式(3), 可得形成 O1—H1Ow 的氢键键能估计值为–18.88 kcal/mol, 属于强氢键范畴。
另外, Ow 和 K1 两原子间电子密度趋于 0, 未找到明确的 BCP 点, 说明不存在典型的 K1―Ow 离子键。但从差分电子密度图(图 5(d))中可以看出, 电子密度在K1 原子处降低; 由 Bader 电荷布居分析可知, 水分子与 O1 原子得到的总电子数大于 H1 原子所失去的电子数, 即 K1 与 Ow 原子之间也存在电子转移现象。在电子局域函数图(图 5(e))中, 钾原子K1 与氧原子 Ow 之间 ELF 值几乎为 0, 代表电子在该处高度离域化, 为离子键特征; 结合钾原子 K1 与水分子中氧原子 Ow 的间距为 3.013Å 且存在着电子转移的现象, 我们推测存在着较弱的闭壳层静电相互作用, 与前人的研究结论[14]相符。
表4 O1—H1Ow键电子密度拓扑分析
Table 4 Topological analysis of electron density of O1—H1Ow bond

参数BD O1—H1Ow0.3320.0670.121–0.017–0.2538
说明: ρ(r)BCP单位为e/bohr3, 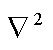 ρ(r)BCP单位为e/bohr5, He(r)BCP单位为hartree/bohr3, BD单位为hartree/e。
ρ(r)BCP单位为e/bohr5, He(r)BCP单位为hartree/bohr3, BD单位为hartree/e。
本文基于第一性原理/分子动力学计算方法, 结合Bader电荷分析、电荷密度、差分电荷密度和电子局域函数等参数, 进行电子密度拓扑分析, 研究单个水分子在 KDP(100)表面的吸附机制, 主要结论如下: 水分子的最佳吸附位点为 H1—K1 桥位, 吸附能为–0.809eV, 说明 KDP 晶体的(100)表面在常温下较易吸附空气中的水分子, 从而产生潮解作用; 末态水分子中的 Ow 原子与 KDP(100)表面磷酸根基团中 H1 原子的间距为 1.578Å, 说明存在过渡闭壳层相互作用, 彼此间形成含共价作用的强氢键, 拟合键能为–18.88kcal/mol; Ow 原子与 KDP(100)表面K1 原子的间距 3.013Å, 两原子之间存在较弱的闭壳层静电相互作用。
参考文献
[1]Bacon G E, Pease R S. A neutron diffraction study of potassium dihydrogen phosphate by fourier synthesis. Proceedings of the Royal Society A: Mathematical, Physical and Engineering Sciences, 1953, 220: 397–421
[2]Yoreo J, Burnham A K, Whitman P K. Developing KH2PO4 and KD2PO4 crystals for the world’s most power laser. International Materials Reviews, 2002, 47(3): 113–152
[3]Fu Y J, Gao Z S, Liu J M, et al. The effects of anionic impurities on the growth habit and optical properties of KDP. Journal of Crystal Growth, 1999, 198(3): 682–686
[4]Fujioka K, Matsuo S, Kanabe T, et al. Optical properties of rapidly grown KDP crystal improved by thermal conditioning. Journal of Crystal Growth, 1997, 181(3): 265–271
[5]滕晓辑. KDP 晶体潮解机理及材料溶解可加工性试验研究[D]. 大连: 大连理工大学, 2007
[6]Yin Y, Zhang Y, Dai Y, et al. Novel magnetor-heological finishing process of KDP crystal by con-trolling fluid-crystal temperature difference to res-train deliquescence. CIRP Annals, 2018, 67(1): 587–590
[7]张秀丽. KDP 晶体潮解抛光的实验研究[D]. 哈尔滨: 哈尔滨工业大学, 2011
[8]Liu Z, Gao H, Guo D. Experimental study on high-efficiency polishing for potassium dihydrogen phos-phate (KDP) crystal by using two-phase air-water fluid. Frontiers of Mechanical Engineering, 2020, 15 (2): 294–302
[9]Mylvaganam K, Zhang L, Zhang Y. Stress-induced phase and structural changes in KDP crystals. Com-putational Materials Science, 2015, 109: 359–366
[10]Zhou G G, Lu G W, Wu C, et al. Study on the struc-ture and surface acoustic wave properties of KDP and ADP crystals. Advanced Materials Research, 2012, 560/561: 66–70
[11]Koval S, Kohanoff J, Migoni R L, et al.Ferroe-lectricity and isotope effects in hydrogen-bonded KDP crystals. Physical Review Letters, 2002, 89(18): 187602
[12]Bromfield T C, Ferré D C, Niemantsverdriet J W. A DFT study of the adsorption and dissociation of CO on Fe (100): influence of surface coverage on the nature of accessible adsorption states. Chemphys-chem, 2010, 6(2): 254–260
[13]Bolton K, Richards T, Mohsenzadeh A. DFT study of the adsorption and dissociation of water on Ni (111), Ni (110) and Ni (100) surfaces. Surface Science: A Journal Devoted to the Physics and Chemistry of Interfaces, 2014, 627: 1–10
[14]Wu Y, Zhang L, Liu Y, et al. Adsorption mechanisms of metal ions on the potassium dihydrogen phosphate (100) surface: a density functional theory-based in-vestigation. Journal of Colloid & Interface Science, 2018, 522: 256–263
[15]Zhang L, Wu Y, Liu Y, et al. DFT study of single water molecule adsorption on the (100) and (101) surfaces of KH2PO4. RSC Advances, 2017, 7(42): 26170–26178
[16]Hartman P. The morphology of zircon and potassium dihydrogen phosphate in relation to the crystal struc-ture. Acta Cryst, 1956, 9(9): 721–727
[17]Vries S D, Goedtkindt P, Bennett S L, et al. Surface atomic structure of KDP crystals in aqueous solution: an explanation of the growth shape. Physical Review Letters, 1998, 80(10): 2229–2232
[18]Hohenberg P, Kohn W. Inhomogeneous electron gas. Physical Review, 1964, 136(3B): B864–B865
[19]Kresse G, Furthmüller J. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Computational Materials Science, 1996, 6(1): 15–50
[20]Kohn W, Sham L J. Self-consistent equations inclu-ding exchange and correlation effects. Physical Re-view, 1965, 140(4A): A1133–A1138
[21]Blochl P E. Projector augmented-wave method. Phys Rev B: Condens Matter, 1994, 50: 17953–17979
[22]Perdew J P, Burke K, Ernzerhof.Generalized gra-dient approximation made simple. Physical Review Letters, 1996, 77(18): 3865–3868
[23]Monkhorst H J, Pack J D. Special points for Brillouin- zone integrations. Physical Review B, 1976, 13(12): 5188–5192
[24]West J. A quantitative X-ray analysis of the structure of potassium dihydrogen phosphate (KH2PO4). Zeits-chrift für Kristallographie—Crystalline Materials, 1930, 74(1): 306–332
[25]Lee K, Murray A, Kong L, et al. A higher-accuracy van der waals density functional. Physical Review B, 2010, 82(8): 081101
[26]Grimme S, Antony J, Ehrlich S, et al. A consistent and accurate ab initio parametrization of density functio-nal dispersion correction (DFT-D) for the 94 elements H-Pu. J Chem Phys, 2010, 132: 154104
[27]Grimme S, Ehrlich S, Goerigk L. Effect of the dam-ping function in dispersion corrected density func-tional theory. J Comput. Chem, 2011, 32(7): 1456–1465
[28]Hoy A R, Bunker P R. A precise solution of the rotation bending Schrdinger equation for a triatomic molecule with application to the water molecule. Journal of Molecular Spectroscopy, 1979, 74(1): 1–8
[29]Bader R F W. Atoms in molecules —a quantum the-ory. Oxford: Clarendon Press, 1990
[30]Becke A D, Edgecombe K. A simple measure of electron localization in atomic and molecular systems. The Journal of Chemical Physics, 1990, 92(9): 5397–5403
[31]Espinosa E, Alkorta I, Elguero J, et al. From weak to strong interactions: a comprehensive analysis of the topological and energetic properties of the electron density distribution involvingX–H⋯F–Ysystems. Journal of Chemical Physics, 2002, 117(12): 5529–5542
[32]Arunan E, De Siraju G R, Klein R A, et al. Definition of the hydrogen bond [EB/OL]. (2011–03–14)[2021–12–01]. http://media.iupac.org/reports/provisional/abs tract11/arunan_prs.pdf
[33]Emamian, S, Lu T, Kruse H, et al. Exploring nature and predicting strength of hydrogen bonds: a correla-tion analysis between atoms-in-molecules descrip-tors, binding energies, and energy components of symmetry-adapted perturbation theory. Journal of Computational Chemistry, 2019, 40(32): 2868–2881
Study on Adsorption of H2O Molecules on KDP(100) Surface Based on DFT the electron density; adsorption
Abstract Based on the calculation method of First-Principles in DFT, the authors carry out the study of the properties of H2O molecules adsorbed on the KDP(100) surface. By topological analysis of the electron density. combining analysis methods such as Bader charge, electron density, electron density difference, electron localization function and other parameters, it is found that the best adsorption site for H2O molecules on KDP(100) surface is H-K bridge site where adsorption energy is –0.809 eV, indicating that the KDP(100) surface can absorb H2O molecules spontaneously. The oxygen atoms in H2O molecules form strong hydrogen bonds O—HOw involving covalent effect, with bond energy –18.88 kcal/mol.
Key words KDP crystal; density functional theory; first-principles molecular dynamics; topological analysis of