原位光反应法合成过氧桥连草酸铀酰配合物
魏文琦 刘春立†
北京分子科学国家研究中心, 放射化学与辐射化学重点学科实验室, 北京大学化学与分子工程学院, 北京 100871; †通信作者, E-mail: liucl@pku.edu.cn
摘要 在室温和实验室灯光照射条件下, 通过原位光化学反应, 分别以乙二胺和二亚乙基三胺为模板, 合成 3 种过氧桥连的草酸铀酰配合物[NH3(CH2)2NH3]3[(UO2)2O2(C2O4)4]·4H2O(1), [NH3(CH2)2NH2(CH2)2NH3]2 [(UO2)2O2(C2O4)4]·2H2O (2)和[NH3(CH2)2NH2(CH2)2NH3]2[(UO2)2O2(C2O4)4] (3)。借助单晶X射线衍射、拉曼光谱(Raman)、粉末 X 射线衍射(PXRD)和热重分析(TGA), 对配合物的结构及其性质进行表征和分析。1 和 2中 U-O2-U 二面角为 180°, 而通常过氧配合物的过氧二面角小于 180°。2 和 3 中过氧键的键长比正常的过氧键长短, 其中 2 更接近超氧键的键长, 但价态和拉曼图谱都表明其为过氧键。
关键词 铀酰配合物; 过氧; 原位; 光反应; 草酸
铀酰过氧配合物对铀矿开采及核燃料后处理具有重要意义。天然的水丝铀矿和变水丝铀矿拥有由铀酰和过氧根配位形成的一维链状结构[1–2], 在地质处置过程中, 由于 α 射线的存在, 放射性废物中的铀可能形成相似结构的过氧化物[1,3]。当铀泄漏到环境中后, 也可能生成多种过氧铀酰配合物[4]。此外, 在回收铀的过程中, 常用的草酸会带来一系列问题, 为了解决这些问题, 通常在工业流程中加入过氧化氢[5]。因此, 关于铀酰过氧配合物的结构及其性质的研究得到越来越多的关注。传统的铀酰过氧配合物是通过加入过氧化物合成的, Burns 研究组[6–9]和 Blanchard 等[5]利用草酸和过氧化氢合成得到双核、多核和笼状结构的配合物。近年来, 一种由光化学反应原位合成而非额外加入过氧根离子的新方法受到研究者的关注。Rose 等[10]于 1994 年首次用原位光反应法合成由 4 个巯基吡啶配位的两个双核铀酰过氧配合物。Thuéry 等[11–12]利用较为复杂的配体合成 3 种由 8 个铀酰围成的笼状配合物。John 等[13]在阳光照射下合成由 6 个 TPPO 配位的铀酰过氧二聚体。Aladzheva 等[14]用自己合成的双齿配体合成了两种新颖的过氧铀酰二聚体, 双齿配体同时与两个铀原子配位形成环状结构。Thangavelu等[15]和 Ridenour 等[16]将吡啶衍生物作为配体, 合成众多以 π—π 相互作用堆积的铀酰过氧二聚体。近期, Jayasinghe 等[17]发现并探讨了不含过氧基团的链状铀酰配合物向过氧铀酰二聚体的转变过程。对于光反应原位产生过氧铀酰配合物, McGrail 等[18]和 Thangavelu 等[15]分别提出两种机理。前者认为过氧根形成是由水或氢氧根与铀酰配位后脱氢所致; 后者则认为氧气与被光致激发的铀酰离子之间发生电子传递产生过氧化氢, 而后过氧化氢与铀酰离子反应生成过氧铀酰配合物。与直接合成方法相比, 原位合成法能够得到独特结构的配合物[19–21]。
本文在室温和实验室灯光照射环境下, 通过原位光反应的方法合成 3 种草酸过氧铀酰配合物, 并借助胺模板平衡电荷及辅助构建结构的功能, 促进晶体的形成。通过单晶 X 射线衍射、粉末 X 射线衍射及拉曼光谱分析和讨论其晶体结构, 发现其中两种配合物的过氧键比正常的过氧键短。同时, 借助热重分析(TGA)研究其热稳定性。
1 实验
1.1 试剂与仪器
试剂包括硝酸铀酰(99%, 中国医药公司北京化学试剂采购供应站)、二水合草酸(99.5%, 西陇化工股份有限公司)、丙二酸钠盐一水合物(99%, 麦克林公司)、乙二胺(99.0%, 西陇化工股份有限公司)、二亚乙基三胺(99%, 九鼎化学公司)和超纯水。所有试剂均为分析纯, 未经进一步提纯。
主要仪器有 XtaLAB PRO 007HF(Mo)单晶 X 射线衍射仪(日本 Rigaku 公司)、X-Pert3 粉末 X 射线衍射仪(荷兰 PANalytical 公司)、元素分析仪(德国Elementar Analysensysteme GmbH 公司)、Q600 热重-差热同步测定仪(美国 TA 公司)、DXRxi 显微拉曼成像光谱仪(美国赛默飞士尔公司)和 FLS980 稳态瞬态荧光/磷光光谱仪(英国爱丁堡公司)。
1.2 合成方法
[NH3(CH2)2NH3]3[(UO2)2O2(C2O4)4]·4H2O(1): 将1.0 mL 草酸(0.1mol/L), 1.0 mL丙二酸钠(0.1 mol/L), 0.2mL 乙二胺(0.5mol/L)和 0.5mL 硝酸铀酰(0.1 mol/L)的水溶液于 10mL 玻璃瓶中混匀(pH=6.7), 置于日光灯下, 每天照射 7 小时。14 天后收取黄绿色针状晶体(1), 用超纯水洗净, 自然晾干, 产率为30%。C, H 和 N 元素(%)分析结果为: C 14.70, H 2.91, N 7.13; 计算结果为: C 14.22, H 3.24, N 7.11。
[NH3(CH2)2NH2(CH2)2NH3]2[(UO2)2O2(C2O4)4]·2H2O(2)和[NH3(CH2)2NH2(CH2)2NH3]2[(UO2)2O2(C2O4)4] (3): 将 1.0 mL 草酸(0.1 mol/L), 1.0 mL 丙二酸钠(0.1 mol/L), 0.2 mL 二亚乙基三胺(0.5 mol/L)和0.5mL 硝酸铀酰(0.1mol/L)的水溶液于 10mL 玻璃瓶中混匀(pH=6.8), 置于日光灯下, 每天照射 7 小时。7 天后收取橘黄色片状晶体 2 和块状晶体 3, 用超纯水洗净, 自然晾干。晶体 2 和 3 可以分离, 但后者产率远低于前者。晶体 2 产率为 72%。C, H 和N 元素(%)分析结果为: C 16.74, H 3.10, N 7.29; 计算结果为: C 16.39, H 3.09, N 7.17。
注意 硝酸铀酰具有 α 放射性, 操作时应避免摄入或沾染。
1.3 表征方法
晶体结构测试和解析 配合物晶体的单晶 X射线衍射数据在恒温 180K 下收集, 入射波长为0.71073Å(MoKα 辐射)。使用 CrysAlisPro 程序进行数据还原和经验吸收校正。用 SHELXT 程序进行结构解析。所有的非氢原子都能够由傅里叶差分图直接确定。骨架氢原子通过几何方法定位, 并使用理论加氢模式连接到母原子。使用 SHELXL 程序, 通过全矩阵技术, 使 F2 的平方偏差之和最小化, 以便完成最终结构精修。配合物 1~3 的 CCDC 号分别为 1888007, 1888009 和 1888010。
热分析 将配合物晶体样品在玛瑙研钵内研细, 取 3~5mg 样品粉末于氧化铝坩埚底铺平。在Q600-SDT 热分析仪上, 采用氮气气氛, 以 5ºC/min的速率升温至 800ºC, 进行热分析测试。采用 TA Universal Analysis 2000 软件进行数据分析和处理。
拉曼光谱分析 将配合物晶体或粉末置于载玻片上, 在 780nm 激光光源下测试。测试参数为: 激光能量为 12.1mW, 曝光时间为 0.5s, 扫描数为 60。
2 结果与讨论
2.1 配合物的晶体结构
晶体数据见表 1~3。
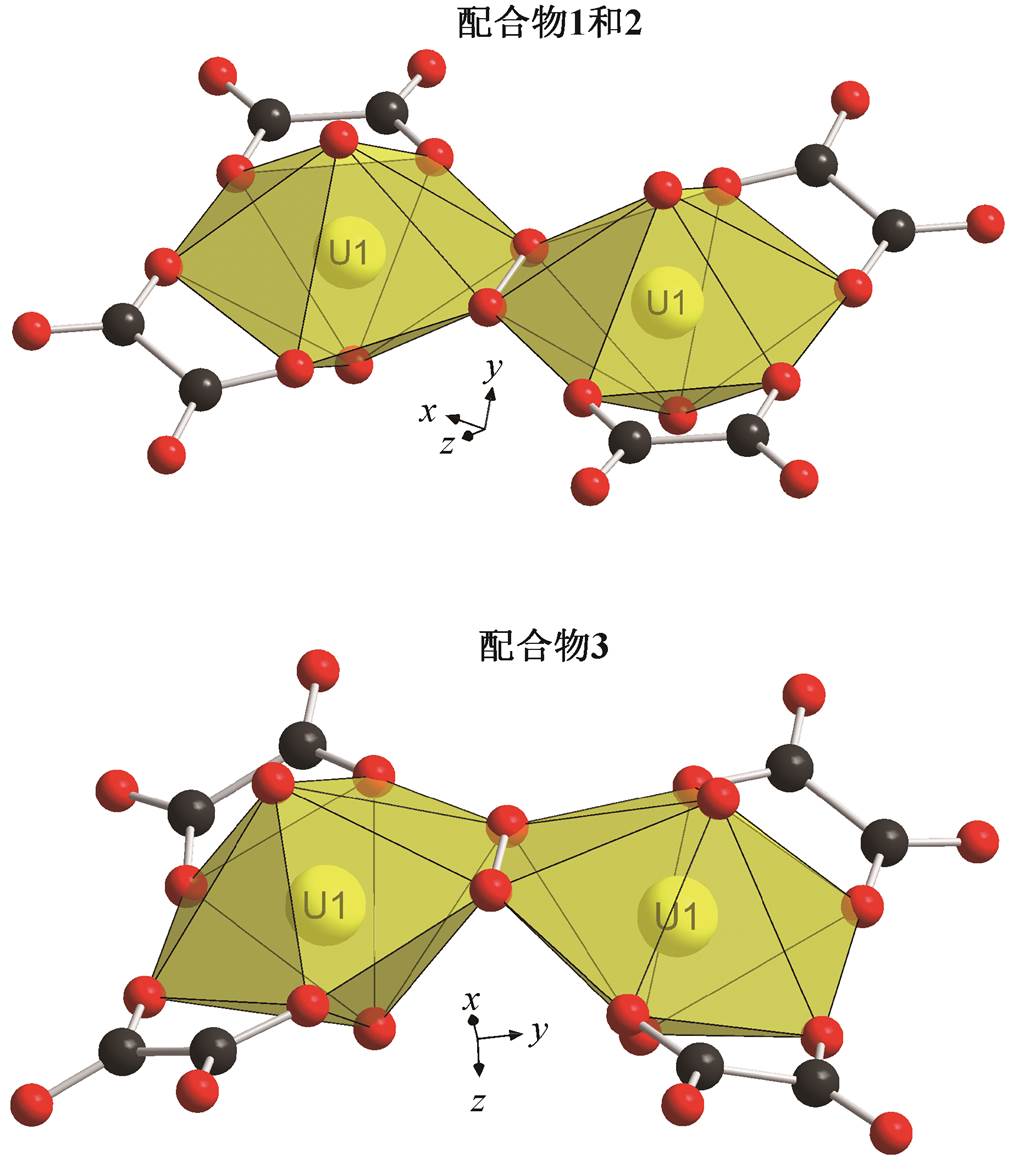
如图 1 和 2 所示, 3 个配合物的骨架均为二聚体, 每个铀酰阳离子均由二齿螯合的草酸离子和四齿螯合的过氧离子配位。铀酰离子的两个轴向氧与6 个配位氧成六角双锥构型。不同的是, 1 和 2 的二聚体成直线型(二面角为 180°), 且相对于过氧根的中点成中心对称, 而 3 的两个六角双锥赤道平面成140.80°夹角。
表1 配合物1~3的晶体学数据
Table 1 Crystal parameters of compounds 1–3
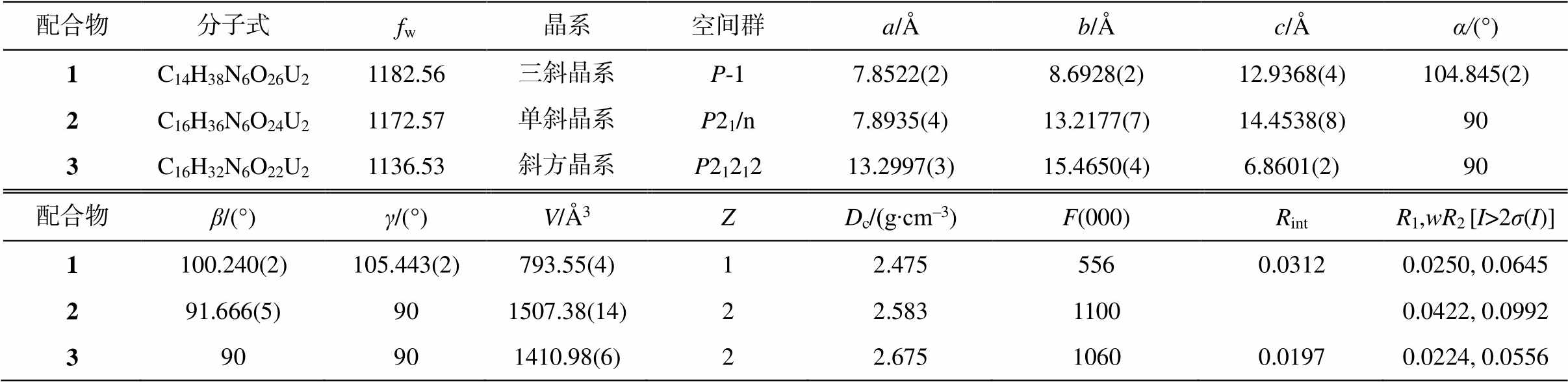
配合物分子式fw晶系空间群a/Åb/Åc/Åα/(°) 1C14H38N6O26U21182.56三斜晶系P-17.8522(2)8.6928(2)12.9368(4)104.845(2) 2C16H36N6O24U21172.57单斜晶系P21/n7.8935(4)13.2177(7)14.4538(8)90 3C16H32N6O22U21136.53斜方晶系P2121213.2997(3)15.4650(4)6.8601(2)90 配合物β/(°)γ/(°)V/Å3ZDc/(g∙cm–3)F(000)RintR1,wR2[I>2σ(I)] 1100.240(2)105.443(2)793.55(4)12.4755560.03120.0250, 0.0645 291.666(5)901507.38(14)22.58311000.0422, 0.0992 390901410.98(6)22.67510600.01970.0224, 0.0556
说明: R1 = Σ||Fo|-|Fc||/Σ|Fo|; wR2={Σ[w(Fo2-Fc2)2]/Σw(Fo2)2}1/2。括号内数字为偏差。下同。
表2 配合物1~3的部分键长(Å)和键角(°)
Table 2 Selected bonds (Å) and angle (°) in compounds 1–3
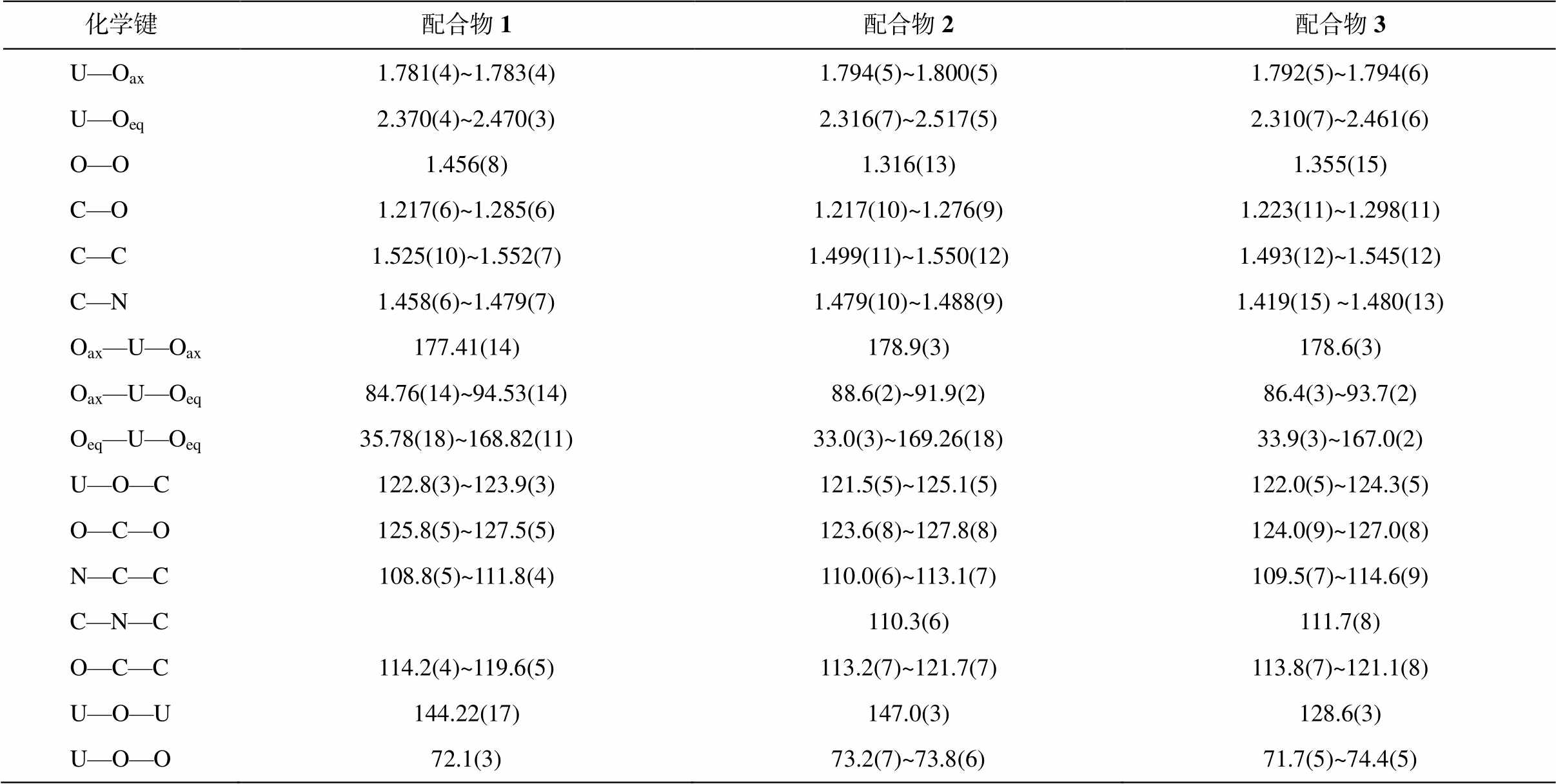
化学键配合物1配合物2配合物3 U—Oax1.781(4)~1.783(4)1.794(5)~1.800(5)1.792(5)~1.794(6) U—Oeq2.370(4)~2.470(3)2.316(7)~2.517(5)2.310(7)~2.461(6) O—O1.456(8)1.316(13)1.355(15) C—O1.217(6)~1.285(6)1.217(10)~1.276(9)1.223(11)~1.298(11) C—C1.525(10)~1.552(7)1.499(11)~1.550(12)1.493(12)~1.545(12) C—N1.458(6)~1.479(7)1.479(10)~1.488(9)1.419(15) ~1.480(13) Oax—U—Oax177.41(14)178.9(3)178.6(3) Oax—U—Oeq84.76(14)~94.53(14)88.6(2)~91.9(2)86.4(3)~93.7(2) Oeq—U—Oeq35.78(18)~168.82(11)33.0(3)~169.26(18)33.9(3)~167.0(2) U—O—C122.8(3)~123.9(3)121.5(5)~125.1(5)122.0(5)~124.3(5) O—C—O125.8(5)~127.5(5)123.6(8)~127.8(8)124.0(9)~127.0(8) N—C—C108.8(5)~111.8(4)110.0(6)~113.1(7)109.5(7)~114.6(9) C—N—C110.3(6)111.7(8) O—C—C114.2(4)~119.6(5)113.2(7)~121.7(7)113.8(7)~121.1(8) U—O—U144.22(17)147.0(3)128.6(3) U—O—O72.1(3)73.2(7)~73.8(6)71.7(5)~74.4(5)
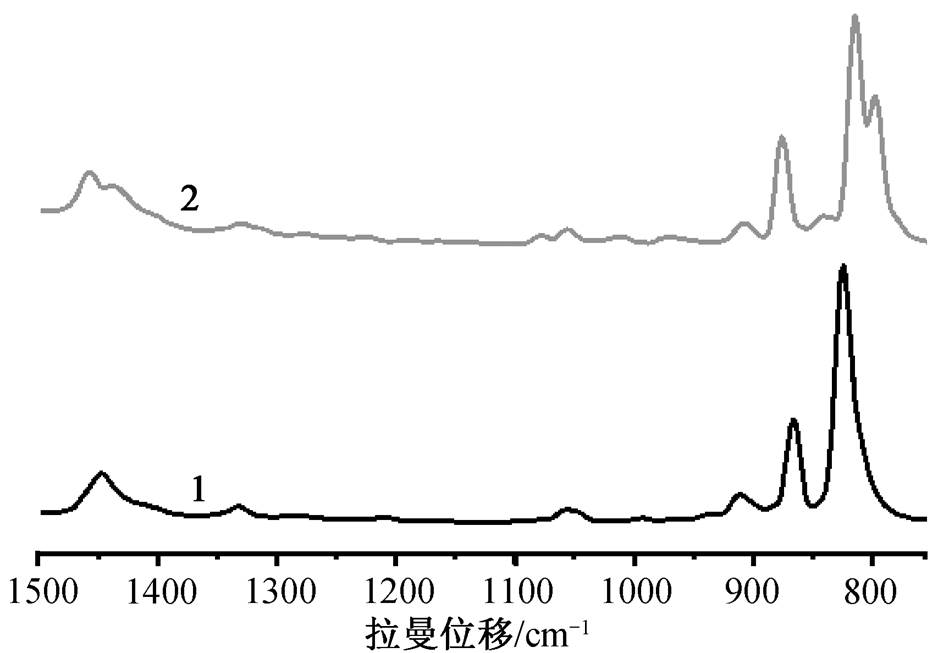
1 的 U=O 和 U—O 键长范围分别是 1.781(4)~ 1.783(4)和 2.370(4)~2.470(3) Å。O—O 键长是 1.456 (8) Å, 在通常报道的过氧键长 1.4~1.5 Å [22]范围内。2 的 U=O 和 U—O 键长范围分别是 1.794(5)~ 1.800(5)和 2.316(7)~2.517(5)Å。其 O—O 键长是1.316(13) Å, 比一般的过氧键长短很多, 更接近超氧键长[23]。3 的 U=O 和 U—O 键长范围分别是 1.792(5)~1.794(6)和 2.310(7)~2.461(6)Å, 与 2 相似的是, 其 O—O 键长是 1.355(15)Å, 也接近超氧键长。但是, 超氧根的价态为−1, 而通过电荷守恒算得到的 2 和 3 中 O—O 的价态为−2 价。此外, 过氧键的伸缩振动峰在拉曼图谱的 800~930cm–1 范围内, 而超氧键的伸缩振动峰则处在 1050~1200cm–1 范围内[6,22,24–26]。图 2 显示, 1 和 2 的 O—O 伸缩振动峰位置十分接近, 1 位于 824 cm–1, 2 位于 840 cm–1, 均位于过氧范围内, 而 2 的 O—O 键长比 1 短, 因此 2 的 O—O 伸缩振动峰位于较高波数, 这也进一步印证了 2 的 O—O 键为过氧键。由于荧光过强, 无法测得 3 的拉曼光谱, 但 3 的结构与 2 极为相似, 所以3也应为过氧配合物。
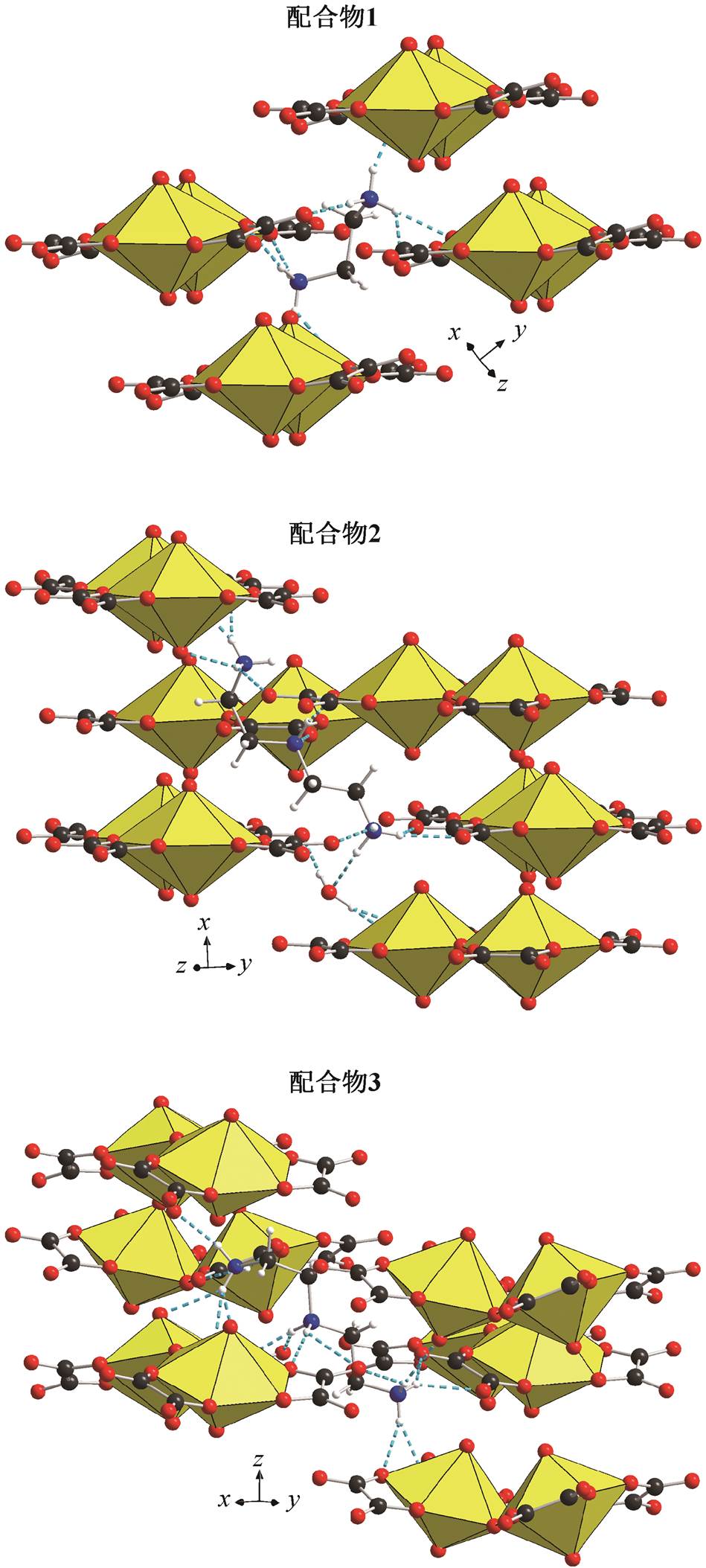
如图 3 所示, 3 种配合物的二聚体均借助与铵根阳离子和晶格水分子(部分未画出)的氢键作用连接在一起, 形成复杂的三维结构。1 中的[NH3(CH2)2 NH3]2+与 4 个二聚体相连, 而 2 和 3 中的[NH3(CH2)2 NH2(CH2)2NH3]3+均与 6 个二聚体相连。水分子填充在二聚体与铵根离子间的空隙中, 进一步提高了结构的强度。二聚体间受空间位阻的影响, 以相互错位的形式平行排列。
表3 配合物1~3的氢键长度(Å)和角度(°)
Table 3 Hydrogen bonds (Å) and angle (°) in compounds 1–3
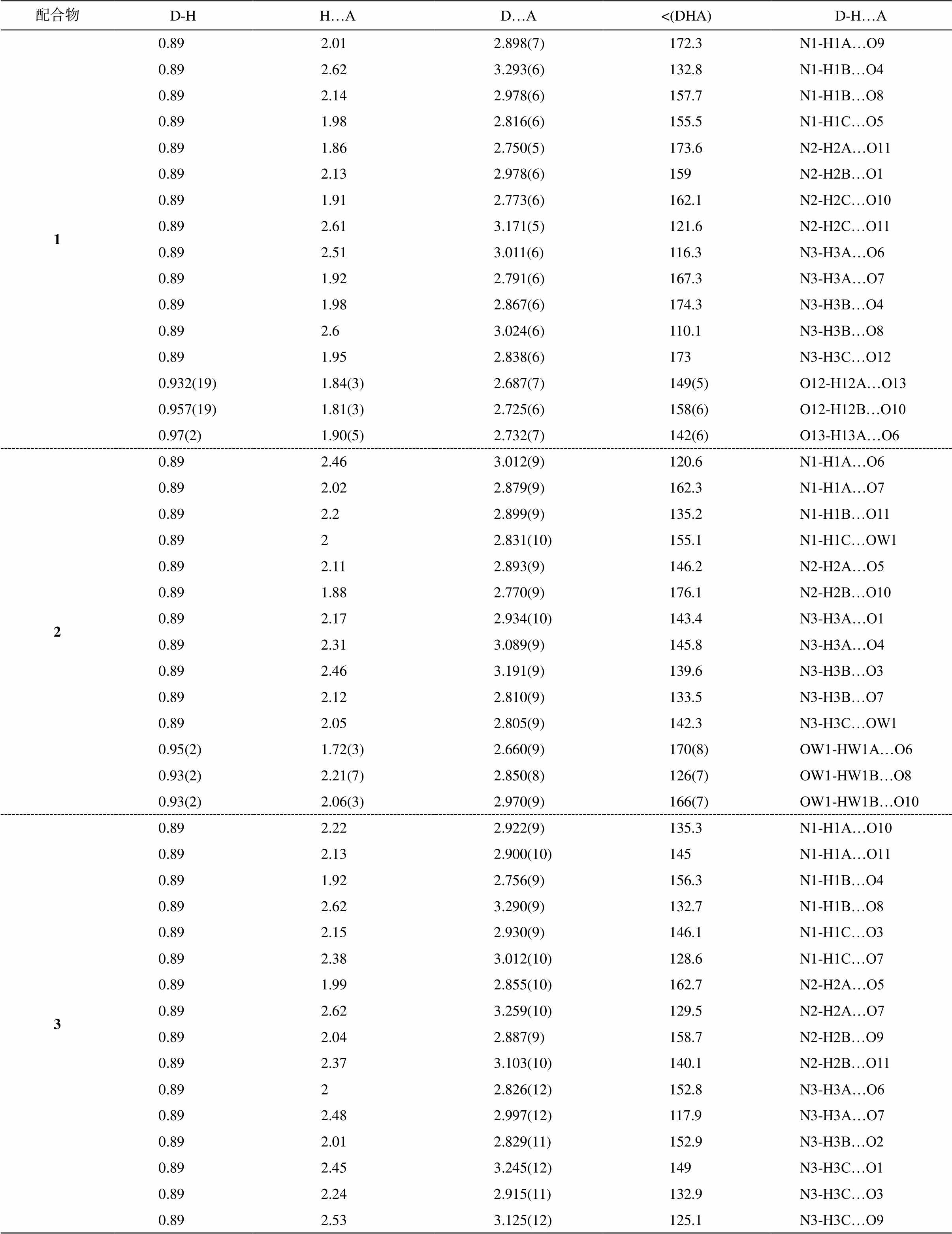
配合物D-H H…AD…A<(DHA)D-H…A 10.892.012.898(7)172.3N1-H1A…O9 0.892.623.293(6)132.8N1-H1B…O4 0.892.142.978(6)157.7N1-H1B…O8 0.891.982.816(6)155.5N1-H1C…O5 0.891.862.750(5)173.6N2-H2A…O11 0.892.132.978(6)159N2-H2B…O1 0.891.912.773(6)162.1N2-H2C…O10 0.892.613.171(5)121.6N2-H2C…O11 0.892.513.011(6)116.3N3-H3A…O6 0.891.922.791(6)167.3N3-H3A…O7 0.891.982.867(6)174.3N3-H3B…O4 0.892.63.024(6)110.1N3-H3B…O8 0.891.952.838(6)173N3-H3C…O12 0.932(19)1.84(3)2.687(7)149(5)O12-H12A…O13 0.957(19)1.81(3)2.725(6)158(6)O12-H12B…O10 0.97(2)1.90(5)2.732(7)142(6)O13-H13A…O6 20.892.463.012(9)120.6N1-H1A…O6 0.892.022.879(9)162.3N1-H1A…O7 0.892.22.899(9)135.2N1-H1B…O11 0.8922.831(10)155.1N1-H1C…OW1 0.892.112.893(9)146.2N2-H2A…O5 0.891.882.770(9)176.1N2-H2B…O10 0.892.172.934(10)143.4N3-H3A…O1 0.892.313.089(9)145.8N3-H3A…O4 0.892.463.191(9)139.6N3-H3B…O3 0.892.122.810(9)133.5N3-H3B…O7 0.892.052.805(9)142.3N3-H3C…OW1 0.95(2)1.72(3)2.660(9)170(8)OW1-HW1A…O6 0.93(2)2.21(7)2.850(8)126(7)OW1-HW1B…O8 0.93(2)2.06(3)2.970(9)166(7)OW1-HW1B…O10 30.892.222.922(9)135.3N1-H1A…O10 0.892.132.900(10)145N1-H1A…O11 0.891.922.756(9)156.3N1-H1B…O4 0.892.623.290(9)132.7N1-H1B…O8 0.892.152.930(9)146.1N1-H1C…O3 0.892.383.012(10)128.6N1-H1C…O7 0.891.992.855(10)162.7N2-H2A…O5 0.892.623.259(10)129.5N2-H2A…O7 0.892.042.887(9)158.7N2-H2B…O9 0.892.373.103(10)140.1N2-H2B…O11 0.8922.826(12)152.8N3-H3A…O6 0.892.482.997(12)117.9N3-H3A…O7 0.892.012.829(11)152.9N3-H3B…O2 0.892.453.245(12)149N3-H3C…O1 0.892.242.915(11)132.9N3-H3C…O3 0.892.533.125(12)125.1N3-H3C…O9
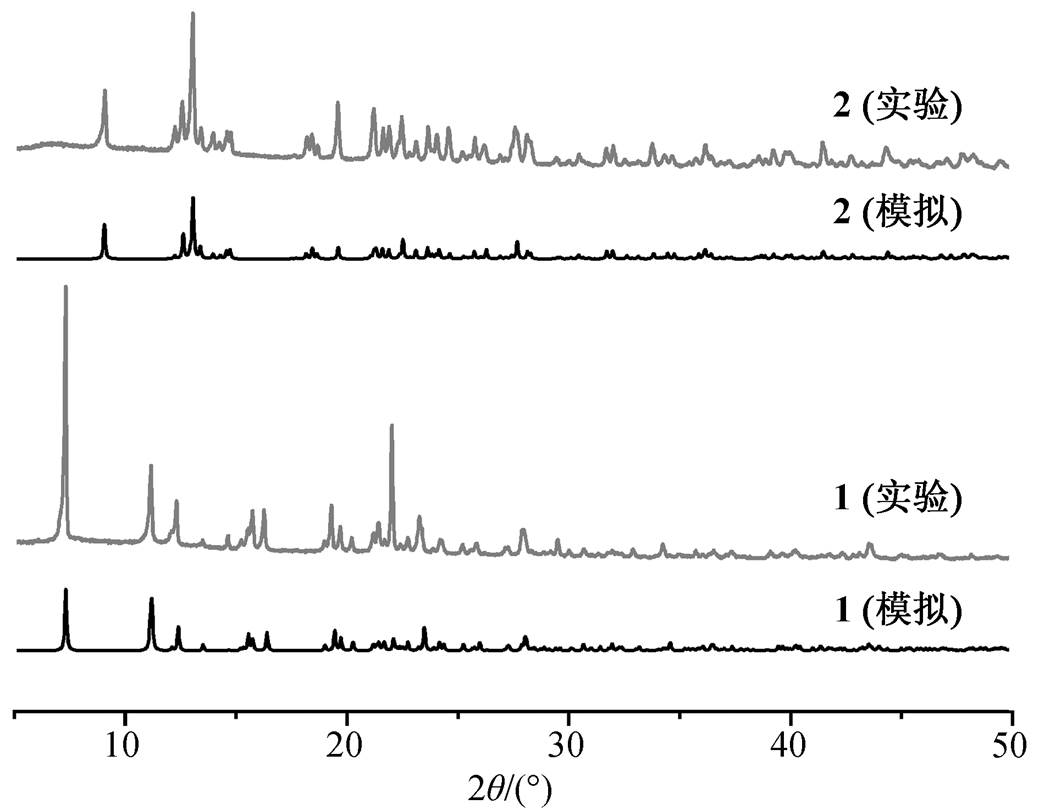
图 4 为配合物 1 和 2 的实验测得和理论拟合粉末衍射图。实验测得的图谱与基于单晶结构数据拟合得到的图谱一致, 因此所得产物纯度很高。
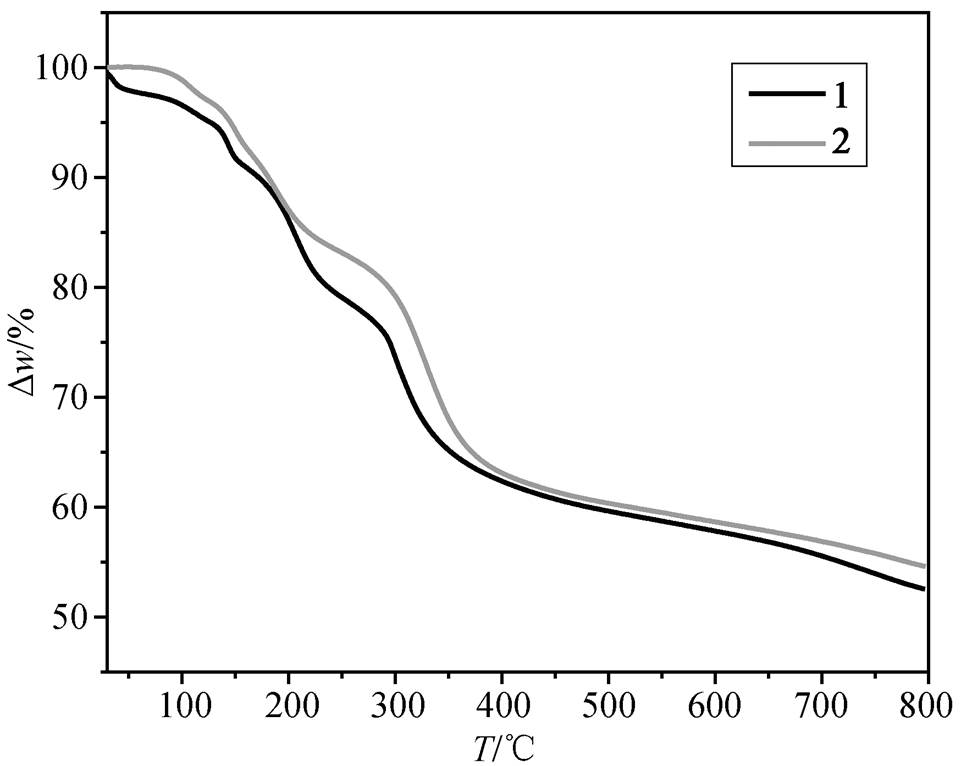
2.2 热分解性质
如图 5 所示, 配合物 1 和 2 的失重曲线非常相似, 主要分为 3 个阶段: 连续失水、有机胺解离和骨架破坏。1 和 2 连续失水过程分别发生于室温至138ºC 和 124ºC, 失重 6.0% (计算值为 6.1%)和 3.0% (计算值为 3.1%)。接着是有机胺解离的过程, 分别至 255ºC 和 297ºC 结束, 失重 15.3% (计算值为15.2%)和 17.4% (计算值为 17.6%)。随后二聚体骨架开始分解, 在氮气气氛下最终产物为 UO2[27–32], 至 800ºC 时残余物比例分别为 52.5%(计算值为 45.7%)和 54.6%(计算值为 46.1%)。失重偏差可能是升温速度较快, 配合物及其中间产物未完全分解所致。因产率过低, 未对3进行热分解测试。
2.3 合成中丙二酸的作用
虽然配合物最终结构中没有丙二酸, 但在晶体制备过程中非常有必要加入丙二酸作为辅助试剂。在没有加丙二酸的样品中, 尽管也能够得到晶体, 但晶体以多晶的形式紧密生长在一起, 无法用于单晶测试。此外, 晶体产量也比加丙二酸的低。
3 结论
本研究在室温和实验室灯光照射条件下, 采用原位光反应合成法, 借助质子化胺模板, 合成 3 种草酸过氧铀酰配合物。3 种配合物的骨架均为二聚体, 其中 1 和 2 中配合物二面角背离过氧化物固有的弯曲特点, 其二面角为 180°。2 和 3 的过氧键长比通常的过氧键长短, 更接近超氧键, 但在价态和拉曼图谱辅证下, 确认它们均为过氧键。二聚体间借助铵离子和水分子, 通过氢键相互连接, 形成三维结构。1 和 2 的热分解曲线表明其历经了连续失水、有机胺解离和骨架破坏 3 个过程, 在惰性气体下, 最终分解为UO2。
参考文献
[1] Kubatko K A H, Helean K B, Navrotsky A, et al. Stability of peroxide-containing uranyl minerals. Sci-ence, 2003, 302: 1191–1193
[2] Burns P C, Hughes K A. Studtite, [(UO2)(O2)(H2O)2] (H2O)2: the first structure of a peroxide mineral. American Mineralogist, 2003, 88(7): 1165–1168
[3] Guo X, Ushakov S V, Labs S, et al. Energetics of metastudtite and implications for nuclear waste alte-ration. Proceedings of the National Academy of Sci-ences of the United States of America, 2014, 111 (50): 17737–17742
[4] Burns P C, Ewing R C, Navrotsky A. Nuclear fuel in a reactor accident. Science, 2012, 335: 1184–1188
[5] Blanchard F, Ellart M, Rivenet M, et al. Role of ammonium ions in the formation of ammonium uranyl peroxides and uranyl peroxo-oxalates. Crystal Growth & Design, 2016, 16(1): 200–209
[6] Qiu J, Vlaisavljevich B, Jouffret L, et al. Cation templating and electronic structure effects in uranyl cage clusters probed by the isolation of peroxide-bridged uranyl dimers. Inorganic Chemistry, 2015, 54(9): 4445–4455
[7] Sigmon G E, Ling J, Unruh D K, et al. Uranyl-peroxide interactions favor nanocluster self-assembly. Journal of the American Chemical Society, 2009, 131(46): 16648–16649
[8] Ling J, Qiu J, Burns P C. Uranyl peroxide oxalate cage and core-shell clusters containing 50 and 120 uranyl ions. Inorganic Chemistry, 2012, 51(4): 2403–2408
[9] Ling J, Wallace C M, Szymanowski J E S, et al. hybrid uranium-oxalate fullerene topology cage clus-ters. Angewandte Chemie-International Edition, 2010, 49(40): 7271–7273
[10] Rose D, Chang Y D, Chen Q, et al. Reactions of uranyl thiolate complexes with molecular oxygen: syntheses and crystal and molecular structures of the uranyl thiolate peroxo species (HNEt3)2[(UO2)2(O2) (SC4N2H3)4] and (HNEt3)[H(UO2)2(O2)(SC4N2H2Me)4]·
Me2CO·0.5Et3N and of the uranyl thiolate oxo cluster (HNEt3)2[(UO2)4(O)2(SC5NH4)6]·Me2CO. Inorganic Chemistry, 1994, 33(23): 5167–5168
[11] Thuéry P, Nierlich M, Baldwin B W, et al. A metal-organic molecular box obtained from self-assembling around uranyl ions. Journal of the Chemical Society-Dalton Transactions, 1999(7): 1047–1048
[12] Thuéry P, Masci B. Self-assembly of an octa-uranate cage complex with a rigid bis-catechol ligand. Supra-molecular Chemistry, 2003, 15(2): 95–99
[13] John G H, May I, Sarsfield M J, et al. The structural and spectroscopic characterisation of three actinyl complexes with coordinated and uncoordinated perrhenate: [UO2(ReO4)2(TPPO)3], [{(UO2)(TPPO)3}2 (μ2-O2)][ReO4]2 and [NpO2(TPPO)4][ReO4]. Dalton Transactions, 2004(5): 734–740
[14] Aladzheva I M, Bykhovskaya O V, Nelyubina Y V, et al. Uranium complexes of cyclic O,O-bidentate ligands with the P-N-P backbone. Inorganica Chimica Acta, 2011, 373(1): 130–136
[15] Thangavelu S G, Cahill C L. Uranyl-promoted pero-xide generation: synthesis and characterization of three uranyl peroxo [(UO2)2(O2)] complexes. Inorganic Chemistry, 2015, 54(9): 4208–4221
[16] Ridenour J A, Cahill C L. Synthesis, structural analysis, and supramolecular assembly of a series of in situ generated uranyl-peroxide complexes with functionalized 2,2'-bipyridine and varied carboxylic acid ligands. New Journal of Chemistry, 2018, 42(3): 1816–1831
[17] Jayasinghe A S, Applegate L C, Unruh D K, et al. Utilizing autoxidation of solvents to promote article the formation of uranyl peroxide materials. Crystal Growth & Design, 2019, 19(3): 1756–1766
[18] McGrail B T, Pianowski L S, Burns P C. Photo-chemical water oxidation and origin of nonaqueous uranyl peroxide complexes. Journal of the American Chemical Society, 2014, 136(13): 4797–4800
[19] Knope K E, Cahill C L. Uranyl triazolate formation via an in situ Huisgen 1,3-dipolar cycloaddition reaction. Crystengcomm, 2011, 13(1): 153–157
[20] Ziegelgruber K L, Knope K E, Frisch M, et al. Hydrothermal chemistry of Th(IV) with aromatic dicarboxylates: new framework compounds and in situ ligand syntheses. Journal of Solid State Chemis-try, 2008, 181(2): 373–381
[21] Knope K E, Cahill C L. Hydrothermal synthesis of a novel uranium oxalate/glycolate via in-situ ligand formation. Inorganic Chemistry, 2007, 46(16): 6607–6612
[22] Cramer C J, Tolman W B, Theopold K H, et al. Variable character of O—O and M—O bonding in side-on (η2) 1:1 metal complexes of O2. Proceedings of the National Academy of Sciences of the United States of America, 2003, 100(7): 3635–3640
[23] Zhang X W, Loppnow G R, McDonald R, et al. {HB(3,5-Me2pz)3}2Sm(η2-O2): first example of a lan-thanide superoxo complex. Journal of the American Chemical Society, 1995, 117(29): 7828–7829
[24] Barraclough C G, Lawrance G A, Lay P A. Cha-racterization of binuclear μ-peroxo and μ-superoxo cobalt(III) amine complexes from Raman spectros-copy. Inorganic Chemistry, 1978, 17(12): 3317–3322
[25] Shibahara T, Mori M. Raman and infrared spectra of μ-O2 dicobalt(III) complexes. Bulletin of the Chemical Society of Japan, 1978, 51(5): 1374–1379
[26] Gubelmann M H, Williams A F. The structure and reactivity of dioxygen complexes of the transition metals. Structure and Bonding, 1983, 55: 1–65
[27] Chugh C A, Sharma A, Sharma A, et al. Kinetics and mechanism of thermal decomposition of uranyl oxalate. Asian Journal of Chemistry, 2011, 23(4): 1865–1866
[28] Schwerdt I J, Olsen A, Lusk R, et al. Nuclear forensics investigation of morphological signatures in the thermal decomposition of uranyl peroxide. Talan-ta, 2018, 176: 284–292
[29] Mihalcea I, Henry N, Bousquet T, et al. Six-fold coordinated uranyl cations in extended coordination polymers. Crystal Growth & Design, 2012, 12(9): 4641–4648
[30] Colmenero F, Bonales L J, Cobos J, et al. Study of the thermal stability of studtite by in situ Raman spectro-scopy and DFT calculations. Spectrochimica Acta Part A—Molecular and Biomolecular Spectroscopy, 2017, 174: 245–253
[31] Donova I, Aleksovska S, Stefov V. Synthesis, charac-terization and thermal decomposition of pyridinium uranyl acetate. Thermochimica Acta, 2000, 348(1/2): 169–174
[32] Cilgi G K, Cetisli H, Donat R. Thermal and kinetic analysis of uranium salts. Journal of Thermal Analysis and Calorimetry, 2014, 115(2): 2007–2020
Synthesis of Oxalate Peroxo-uranyl Complex in situ by Photoreaction
WEI Wenqi, LIU Chunli†
Beijing National Laboratory for Molecular Sciences, Fundamental Science Laboratory on Radiochemistry and Radiation Chemistry, College of Chemistry and Molecular Engineering, Peking University, Beijing 100871; † Corresponding author, E-mail:liucl@pku.edu.cn
Abstract [NH3(CH2)2NH3]3[(UO2)2O2(C2O4)4]·4H2O (1), [NH3(CH2)2NH2(CH2)2NH3]2[(UO2)2O2(C2O4)4]·2H2O (2) and [NH3(CH2)2NH2(CH2)2NH3]2[(UO2)2O2(C2O4)4] (3) were synthesized in situ under room temperature and laboratory lamp condition, using 1,2-diaminoethane and 2,2'-diaminodiethylamine separately as templates. Structures and properties were characterized by single-crystal X-ray diffraction, Raman spectra, powder X-ray diffraction (PXRD) and thermogravimetric analysis (TGA).The dihedral angle of U-O2-U in 1 and 2 is 180°, which deviates from the natural angle of peroxide. The peroxide bond length of 2 and 3 is shorter than that of the reported peroxide, and the bond lenth of 2is close to superoxide bond length. However, both valence and Raman spectra indicated that2 and 3are peroxide bonds.
Key words uranyl complex; peroxide; in situ; photoreaction; oxalic acid
doi: 10.13209/j.0479-8023.2020.016
国家自然科学基金(11475008, U1530112, U1730245)资助
收稿日期: 2019-03-15;
修回日期: 2019-06-06