
图1 长江下游采样点分布
Fig. 1 Sampling sites in the lower Yangtze River
摘要 为探索河流系统中厌氧氨氧化细菌的分布情况, 于2014年春秋两季采集长江下游6个断面的水体和沉积物样品, 通过宏基因组测序方法, 根据优化序列与自建厌氧氨氧化细菌标记基因hzsA, hzsB, hzsC, hdh数据集的比对结果, 计算各基因相对丰度。结果表明, 水体溶解氧浓度较高导致厌氧氨氧化细菌标记基因丰度极低, 而沉积物中各基因相对丰度较高, 平均值分别为4.540×10−10, 4.939×10−10, 4.333×10−10和2.859×10−10。随着温度升高, 秋季沉积物中各基因相对丰度显著高于春季。大通、南京和徐六泾沉积物中各基因相对丰度较高, 这与人类活动扰动的增强以及入海口盐度升高有关。物种分类鉴定结果表明, 长江下游沉积物中厌氧氨氧化细菌在属水平上以Candidatus Brocadia和Candidatus Jettenia为主。厌氧氨氧化细菌标记基因相对丰度与NO2−-N, NO3−-N和NH4+-N等理化因子相关, 并且, 由于NO2−-N浓度远远低于NH4+-N, 因此NO2−-N浓度是长江下游沉积物中厌氧氨氧化细菌生长的限制性因子。
关键词 长江下游; 厌氧氨氧化细菌; 标记基因; 物种组成
厌氧氨氧化(anaerobic ammonium oxidation, anammox)指厌氧氨氧化细菌在缺氧条件下以NO2−为电子受体将NH4+氧化为N2的过程。目前发现的厌氧氨氧化细菌主要隶属于浮霉菌门Planctomyce-tes下的6个属: Candidatus Brocadia, Candidatus Kuenenia, Candidatus Scalindua, Candidatus Jettenia, Candidatus Anammoxoglobus和Candidatus Anam-moximicrobium[1]。厌氧氨氧化细菌主要存在于具有较大无机氮通量的缺氧环境中, 如农业土壤[2]、湖泊湿地[3]、海洋沉积物[4]和含氮废水[5]等。
河流沿岸是人类聚集度较高和农业活动频繁的区域, 受点源和面源氮污染的影响, 河流中可能存在厌氧氨氧化细菌。Zhang等[6]通过16S rRNA基因扩增子测序, 在江苏省新沂河沉积物中检测到厌氧氨氧化细菌Candidatus Brocadia和Candidatus Scali-ndua。Hu等[7]通过16S rRNA基因扩增子测序和定量PCR等方法, 在钱塘江沉积物中检测到CandidatusBrocadia, Candidatus Kuenenia和Candidatus Scalin-dua, 且以前两者为主, 丰度主要受有机和无机碳含量的影响。Sun等[8]通过16S rRNA基因扩增子测序和定量PCR等方法, 在广东省东江沉积物中检测到Candidatus Brocadia, 相关性分析表明亚硝酸盐浓度是影响厌氧氨氧化细菌丰度的关键因子。
由于厌氧氨氧化细菌很难纯培养, 已有研究主要通过对目标序列(如16S rRNA)使用特异性引物扩增提取后测序分析, 而PCR扩增的偏好性和引物对目标序列的覆盖度可能造成对基因丰度的低估, 甚至检测不到低丰度的基因[9]。宏基因组测序则对样品中全部DNA构建基因文库进行测序, 能够更全面地反映环境样品中的微生物组成。研究表明, 编码联氨合成酶(hydrazine synthase, HZS)3 个亚基的基因hzsA, hzsB, hzsC和编码联氨脱氢酶(hydrazine dehydrogenase, HDH)的基因hdh常作为厌氧氨氧化细菌的标记基因[10–11]。
长江下游地区是中国人口密度最大和城市化程度最高的地区之一, 接纳长江流域大量的人为氮排放, 包括农业活动、渔业、城市和工业污水排放等[12],因此对长江下游地区厌氧氨氧化细菌丰度及时空分布规律进行研究, 对揭示长江下游氮转化过程有重要意义。本研究采用宏基因组测序方法, 根据hzsA, hzsB, hzsC和hdh基因相对丰度, 间接表征厌氧氨氧化细菌在长江下游各采样点的丰度, 进而分析厌氧氨氧化细菌的主要物种组成和影响因素。
为研究长江下游厌氧氨氧化细菌的丰度及沿程分布状况, 在长江下游干流布设6个采样点(图1), 于2014年春季(3月)和秋季(10月)采集微生物水体和沉积物样品。在每个采样点用5L无菌聚乙烯瓶采集10 L水样, 于24小时内用孔径0.22 μm的聚碳酸酯滤膜(Millipore, 美国)过滤完毕, 滤膜贮藏于−80℃冰箱。沉积物样品为对应水样岸边0.5m水深下的表层沉积物样品, 为避免沉积物的异质性扰动, 根据实际条件, 每隔50m采集1~3个样品, 装于50mL灭菌离心管中, −80℃冷冻保存。
图1 长江下游采样点分布
Fig. 1 Sampling sites in the lower Yangtze River
采样点基础信息及理化指标是影响厌氧氨氧化细菌丰度及沿程分布的关键因子。本研究在采集微生物样品的同时, 同步记录采样点的经纬度、海拔和距河口距离等基础信息, 测定溶解氧、总氮、氨氮(NH4+-N)、亚硝酸盐氮(NO2−-N)、硝酸盐氮(NO3−-N)、总有机碳和总磷等的浓度及水温和pH, 所有指标测定方法均参照相应的国家标准。
取两张滤膜置于2mL灭菌离心管中, 液氮速冻后捣碎, 使用FastDNA® SPIN Kit for soil试剂盒(MP Biomedicals, 美国)进行水样微生物DNA提取。沉积物样品于室温下融化, 离心后取0.5~1.0g 样品至2 mL灭菌离心管, 采用同样的方法提取DNA。每个样品进行3~5次DNA提取, 并进行混合组成该样品的DNA样本。之后使用0.8%的琼脂糖凝胶电泳检测DNA的完整性, 使用NanoDrop 2000(Thermo Fi-sher Scientific, 美国)检测DNA的浓度和纯度。
DNA样品置于干冰中, 运输至上海美吉生物医药科技有限公司, 根据Illumina Hiseq 4000测序平台 的标准流程进行双端测序。测序长度为2×150bp, 采用Sickle[13]和NGS QC Toolkit[14]对下机原始数据进行质量控制, 去除低质量条带, 得到优化序列(clean reads)。之后使用 IDBA-UD[15]进行拼接, 使用MetaGeneMark[16]进行开放阅读框(open reading frame, ORF)预测。从 NCBI Protein 数据库中分别下载厌氧氨氧化细菌 hzsA, hzsB, hzsC, hdh 基因氨基酸序列构建数据集, 将优化序列与各数据集用UBLAST[17]和BLASTX[18]进行比对, 依据identity ≥ 80%且alignment length ≥ 75 bp的标准, 筛选出有效的比对结果。用该比对结果除以样品优化序列总数和基因最大序列长度, 即得到各类基因的相对丰度。将ORF氨基酸序列与各基因数据集进行DIAMOND[19]比对, 获得的有效比对序列(identity≥ 80%)与参考序列进行MAFFT[20]对齐, 之后使用FastTree[21]构建系统发育树。
使用SPSS 18.0软件, 对4类基因之间、基因相对丰度与理化指标之间的相关性进行分析。使用Gephi0.9.2[22]构建各基因相对丰度与理化因子相关性网络结构图。
春季和秋季各监测断面的hzsA, hzsB, hzsC和hdh基因相对丰度如图2所示。可以看出, 春季水体(water spring, WS)和秋季水体(water autumn, WA)中hzsA, hzsB, hzsC和hdh基因相对丰度极低, 平均值分别为(0.144±0.110)×10−10, (0.102±0.111)×10−10, (0.136 ±1.138)×10−10和(0.053±0.067)×10−10, 且沿程变化趋势不明显。这是因为厌氧氨氧化细菌是厌氧细菌, 适宜生存的溶解氧浓度约为0.065mg/L[23], 而本研究监测断面水体溶解氧浓度的平均值为9.0±1.4 mg/L, 为适宜溶解氧浓度的100倍以上, 不利于厌氧氨氧化细菌的生长。然而, 在春季沉积物(sedi-ment spring, SS)和秋季沉积物(sediment autumn, SA)中显著地检测到各标记基因, 远大于水体中厌氧氨氧化细菌各基因丰度, 这与沉积物较低的溶解氧含量有关。
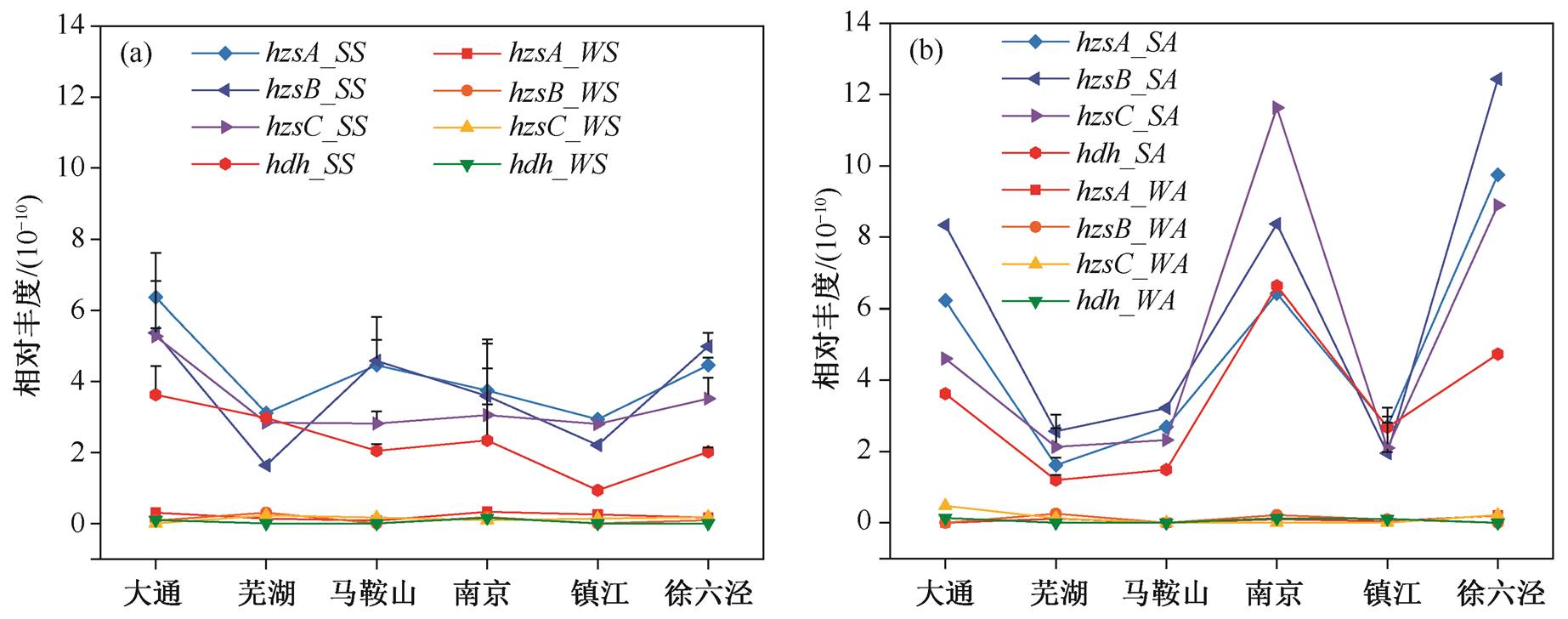
图2 2014年春季(a)和秋季(b)的hzsA, hzsB, hzsC和hdh基因相对丰度
Fig. 2 Relative abundance of hzsA, hzsB, hzsC, hdh genes in spring (a) and autumn (b) in 2014
从季节变化来看, 秋季沉积物中各基因相对丰度平均值高于春季沉积物, 这是因为秋季水温平均值为23.28±0.39℃, 远高于春季水温10.18±0.70℃。Laureni等[24]利用厌氧氨氧化细菌处理城市污水的实验室研究表明, 温度为29℃时, 厌氧氨氧化细菌倍增时间为18天, 去除氮的速率为465mgN/(L·d); 当温度为12.5℃时, 厌氧氨氧化细菌倍增时间增加至79天, 去除氮的速率降低至46mgN/(L·d)。To-maszewski等[25]的研究也发现, 厌氧氨氧化细菌适宜的生长温度为30~40℃。本研究秋季水温更接近厌氧氨氧化细菌的最佳适宜温度, 因此, hzsA, hzsB, hzsC和hdh基因的相对丰度均是秋季大于春季。
从沿程变化来看, 秋季沉积物中, 4类基因的相对丰度沿程变化显著且趋势一致, 基因相对丰度较高的断面为大通、南京和徐六泾。调查表明, 大通断面附近有较多的支流汇入, 北岸为农业种植区, 南岸为工业区和居民住宅区, 输入的营养物质为厌氧氨氧化细菌的生长提供了有利条件; 南京断面附近分布着金陵石化、金陵石化复肥厂和南京金星石化等工业企业, 受雨水冲刷的影响, 采样点沉积物中氮素浓度较高, 且温度适宜, 促进厌氧氨氧化细菌的生长; 徐六泾断面为长江的入海口, 淡水和海水交汇, 营养物质丰富, 盐度较高, 厌氧氨氧化细菌的丰度随之升高[26]。相比较而言, 春季沉积物中各基因的沿程变化小得多, hzsc和hdh基因甚至看不出明显的沿程变化, 这与春季雨水较少, 人类活动扰动引起的生源物质汇入较少有关, 且春季温度较低, 不利于厌氧氨氧化细菌的繁殖。
为进一步揭示各基因相对丰度之间的相关性, 对4类基因丰度进行Pearson相关性分析, 结果如表1所示。可以看出, 4类标记基因两两之间均显著正相关, 相关系数为0.8615~0.9590, 显著性水平小于0.001。这是因为4类基因均位于厌氧氨氧化细菌的基因组上[27], 在宏基因组测序过程中同时被测序, 其相对丰度与厌氧氨氧化细菌的数量和丰度呈正相关关系。
表1 hzsA, hzsB, hzsC和hdh基因相对丰度的Pearson相关性分析
Table 1 Pearson correlation analysis of relative abundances of hzsA, hzsB, hzsC and hdh genes

基因hzsAhzsBhzsChdh hzsA 1.00000.9590***0.8781***0.9059*** hzsB0.9590*** 1.00000.8668***0.8615*** hzsC0.8781***0.8668*** 1.0000 0.9301*** hdh0.9059***0.8615***0.9301*** 1.0000
注: ***表示在0.001水平(双侧)上显著相关。
为确定长江下游沉积物中所含厌氧氨氧化细菌的种类, 根据ORF比对各基因集的结果构建系统发育树, 如图3所示。结合4类基因系统发育分析的结果可以看到, 大通春季和秋季沉积物中均含Candi-datus Brocadia和Candidatus Jettenia; 马鞍山春季沉积物中含Candidatus Jettenia, 秋季沉积物中含Candi-datus Brocadia; 南京采样点春季沉积物中含Can-didatus Brocadia, 秋季沉积物中含Candidatus Bro-cadia和Candidatus Jettenia; 镇江秋季沉积物中含Candidatus Brocadia; 徐六泾春季和秋季沉积物中均含Candidatus Brocadia。考虑到沉积物的相对稳定性, 推测长江下游沉积物中所含厌氧氨氧化细菌以Candidatus Brocadia和Candidatus Jettenia为主。
沉积物中厌氧氨氧化细菌基因丰度极易受各类理化因子的影响。将Spearman相关性分析中显著性<0.05的因子提取出来, 进行网络结构分析, 结果如图4所示。图形的模块化参数为0.529, 说明该网络结构图是有效的[28]。如前所述, hzsA, hzsB, hzsC和hdh基因相对丰度之间存在相关性。此外, 它们还与NH4+-N和NO3−-N浓度显著相关, 后两者是厌氧氨氧化细菌直接或间接利用的营养物质。
研究表明, 只有NH4+和NO2−同时存在时, 厌氧氨氧化细菌才能进行厌氧氨氧化反应, 且NH4+与NO2−的理论化学计量比为1:1.32[29]。本研究采集的沉积物样品中, NH4+-N浓度范围为0.245~22.130 mg/L, 平均值为8.912±7.962mg/L; NO2−-N的浓度范围为0.040~0.294mg/L, 平均值为0.125±0.085 mg/L。与NH4+-N相比, NO2−-N处于严重不足的状态。Sun等[8]的研究表明, 东江沉积物的NH4+-N浓度为39.26~287.63mg/L, NO2−-N的浓度为0.15~0.90 mg/L, NO2−-N是影响厌氧氨氧化细菌丰度的关键因子。与之类似, 本研究对各基因相对丰度与NO2−-N浓度进行Spearman偏相关分析(表2), 结果表明, hzsA, hzsB, hzsC和hdh基因相对丰度均与NO2−-N浓度正相关, 说明长江下游沉积物中, NO2−-N浓度是厌氧氨氧化细菌生长的限制性因子。
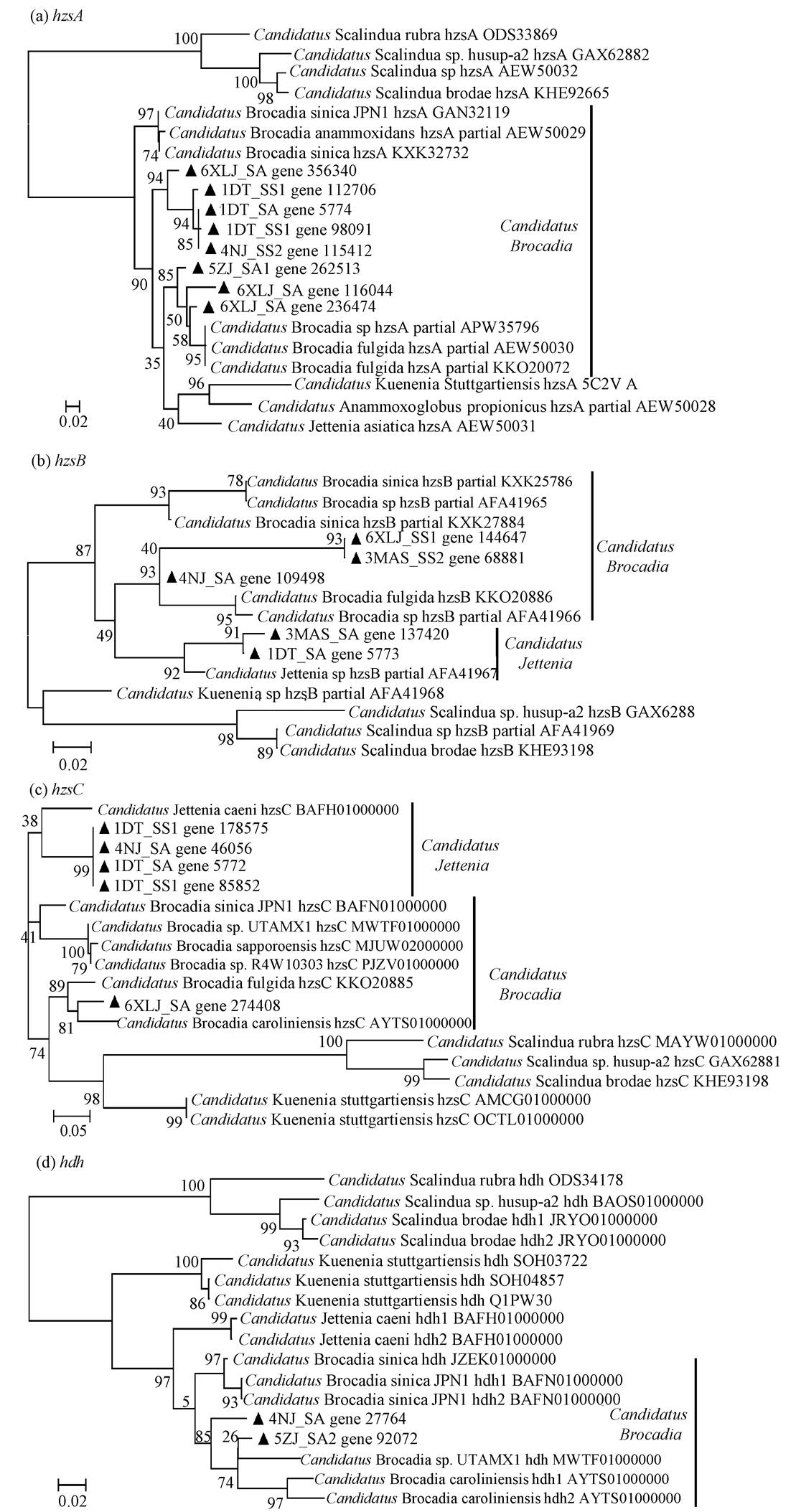
图3 沉积物样品中hzsA, hzsB, hzsC和hdh基因系统发育树
Fig. 3 Phylogenetic tree of hzsA, hzsB, hzsC and hdh gene sequences recovered from sediment samples
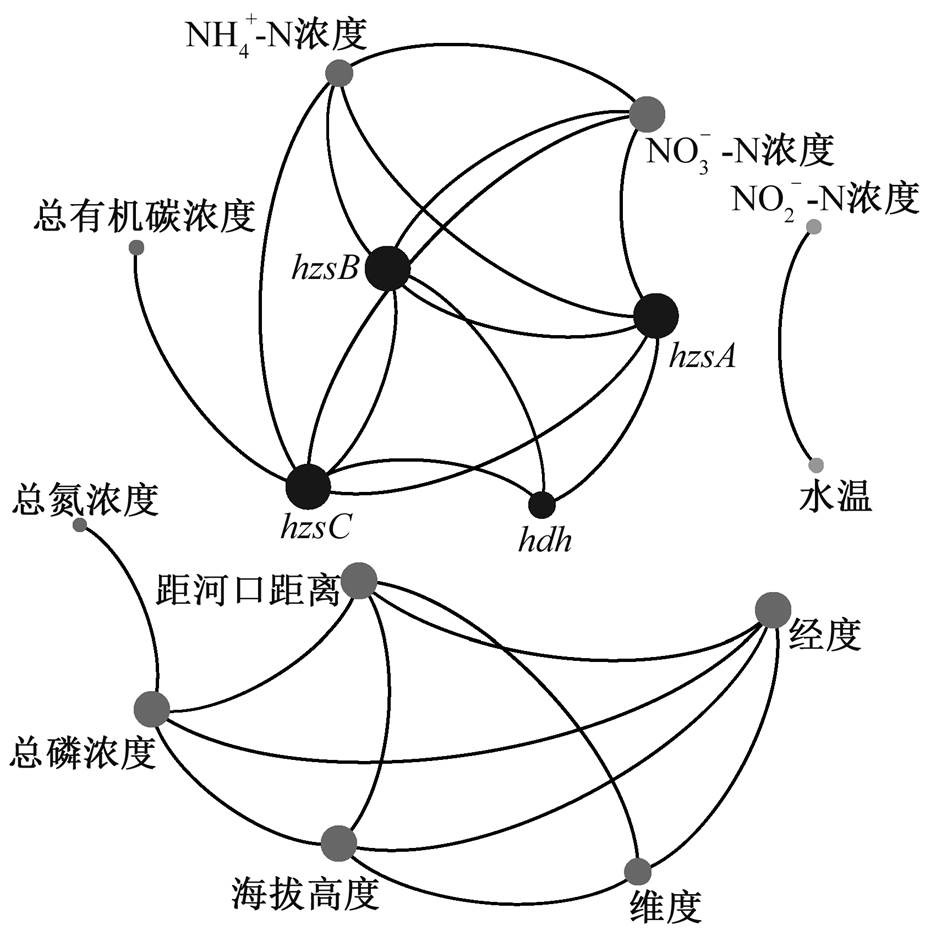
图4 hzsA, hzsB, hzsC和 hdh基因相对丰度与理化因子相关性网络结构
Fig. 4 Correlation network of hzsA, hzsB, hzsC, hdh relative abundances and environmental factors
表2 hzsA, hzsB, hzsC和hdh基因相对丰度与NO2−-N浓度偏相关分析
Table 2 Partial correlation analysis of hzsA, hzsB, hzsC, hdh relative abundances with NO2−-N concentration

基因NO2−-N浓度 Spearman相关性显著性(双侧) hzsA0.9350.065 hzsB0.8300.170 hzsC0.9920.008 hdh0.8880.112
本文利用宏基因组高通量测序研究长江下游(大通至徐六泾)水体和沉积物厌氧氨氧化细菌的相对丰度及其影响因素, 得到如下主要结论。
1)受溶解氧浓度影响, 长江下游水体中hzsA, hzsB, hzsC和hdh4类厌氧氨氧化细菌标记基因丰度极低, 而沉积物各基因相对丰度较高。随着温度的升高, 秋季沉积物中各基因相对丰度平均值高于春季。
2)从沿程变化来看, 大通和南京各基因相对丰度较高, 这与沿岸人类活动的显著影响有关, 徐六泾断面基因丰度高则与入海口盐度较高的特殊条件有关。
3)物种分类鉴定结果表明, 长江下游沉积物中厌氧氨氧化细菌以Candidatus Brocadia和Candida-tus Jettenia为主。
4) hzsA, hzsB, hzsC和hdh基因相对丰度与NO2−-N, NO3−-N和NH4+-N浓度相关, 且由于NO2−浓度远远低于NH4+-N浓度, NO2−-N浓度是长江下游沉积物中厌氧氨氧化细菌生长的限制性因子。
参考文献
[1]胡倩怡, 郑平, 康达. 厌氧氨氧化菌的种类、特性与检测. 应用与环境生物学报, 2017, 23(2): 384–391
[2]Humbert S, Tarnawski S, Fromin N, et al. Molecular detection of anammox bacteria in terrestrial ecosys-tems: distribution and diversity. The ISME Journal, 2010, 4(3): 450–454
[3]王衫允, 祝贵兵, 曲冬梅, 等. 白洋淀富营养化湖泊湿地厌氧氨氧化菌的分布及对氮循环的影响. 生态学报, 2012, 32(21): 6591–6598
[4]Dalsgaard T, Thamdrup B, Canfield D E. Anaerobic ammonium oxidation (anammox) in the marine envi-ronment. Research in Microbiology, 2005, 156(4): 457–464
[5]Van D U, Jetten M S, van Loosdrecht M C. The SHARON®-Anammox® process for treatment of am-monium rich wastewater. Water Science and Techno-logy, 2001, 44(1): 153–160
[6]Zhang Y, Ruan X H, Hj O D C, et al. Diversity and abundance of aerobic and anaerobic ammonium-oxidizing bacteria in freshwater sediments of the Xinyi River (China). Environmental Microbiology, 2007, 9(9): 2375–2382
[7]Hu Baolan, Shen Lidong, Zheng Ping, et al. Distribu-tion and diversity of anaerobic ammonium-oxidizing bacteria in the sediments of the Qiantang River. Environmental Microbiology Reports, 2012, 4(5): 540–547
[8]Sun Wei, Xu Meiying, Wu Weimin, et al. Molecular diversity and distribution of anammox community in sediments of the Dongjiang River, a drinking water source of Hong Kong. Journal of Applied Microbio-logy, 2014, 116(2): 464–476
[9]Amano T, Yoshinaga I, Okada K, et al. Detection of anammox activity and diversity of anammox bacteria-related 16S rRNA genes in coastal marine sediment in Japan. Microbes and Environments, 2007, 22(3): 232–242
[10]Bhattacharjee A S, Wu S, Lawson C E, et al. Whole-community metagenomics in two different anammox configurations: process performance and community structure. Environmental Science & Technology, 2017, 51(8): 4317–4327
[11]Harhangi H R, Le Roy M, van Alen T, et al. Hydrazine synthase, a unique phylomarker with which to study the presence and biodiversity of anammox bacteria. Applied and Environmental Microbiology, 2012, 78(3): 752–758
[12]Hou Lijun, Liu Min, Xu Shiyuan, et al. The diffusive fluxes of inorganic nitrogen across the intertidal sediment-water interface of the Changjiang Estuary in China. Acta Oceanologica Sinica, 2006, 25(3): 48–57
[13]Joshi N, Fass J. Sickle: a sliding-window, adaptive, quality-based trimming tool for fastq files (Version 1.33) [EB/OL]. (2011) [2018–04–01]. https://github.com/najoshi/sickle
[14]Patel R K, Jain M. NGS QC toolkit: a toolkit for quality control of next generation sequencing data. Plos One, 2012, 7(2): e30619
[15]Yu Peng, Leung H C, Yiu S M, et al. IDBA-UD: a denovo assembler for single-cell and metagenomic sequencing data with highly uneven depth. Bioinfor-matics, 2012, 28(11): 1420–1428
[16]Zhu W H, Lomsadze A, Borodovsky M. Ab initio gene identification in metagenomic sequences. Nucleic Acids Research, 2010, 38(12): e132
[17]Edgar R C. Search and clustering orders of magnitude faster than BLAST. Bioinformatics, 2010, 26(19): 2460–2461
[18]Lobo I. Basic local alignment search tool (BLAST). Journal of Molecular Biology, 2012, 215(3): 403–410
[19]Buchfink B, Xie Chao, Huson D H. Fast and sensitive protein alignment using DIAMOND. Nature Methods, 2015, 12(1): 59–60
[20]Katoh K, Standley D M. MAFFT multiple sequence alignment software version 7: improvements in per-formance and usability. Molecular Biology and Evo-lution, 2013, 30(4): 772–780
[21]Price M N, Dehal P S, Arkin A P. FastTree 2-approximately maximum-likelihood trees for large alignments. Plos One, 2010, 5(5): e9490
[22]Jacomy M, Bastian M, Heymann S. Gephi: an open source software for exploring and manipulating networks // Proceedings of the Third International Conference on Weblogs and Social Media. San Jose, 2009: doi: 10.13140/2.1.1341.1520
[23]Strous M, Van G E, Kuenen J G, et al. Effects of aerobic and microaerobic conditions on anaerobic ammonium-oxidizing (anammox) sludge. Applied and Environmental Microbiology, 1997, 63(6): 2446–2448
[24]Laureni M, Weissbrodt D G, Szivák I, et al. Activity and growth of anammox biomass on aerobically pre-treated municipal wastewater. Water Research, 2015, 80: 325–336
[25]Tomaszewski M, Cema G, Ziembińskabuczyńska A. Influence of temperature and pH on the anammox process: a review and meta-analysis. Chemosphere, 2017, 182: 203–214
[26]Koop-Jakobsen K, Giblin A E. Anammox in tidal marsh sediments: the role of salinity, nitrogen loa-ding, and marsh vegetation. Estuaries and Coasts, 2009, 32(2): 238–245
[27]Speth D R, In’t Zandt M H, Simon G C, et al. Genome-based microbial ecology of anammox gra-nules in a full-scale wastewater treatment system. Nature Communications, 2016, 7: 11172
[28]Newman M E. Modularity and community structure in networks // APS March Meeting. Baltimore: American Physical Society, 2006: 8577–8582
[29]Strous M, Heijnen J J, Kuenen J G, et al. The sequen-cing batch reactor as a powerful tool for the study of slowly growing anaerobic ammonium-oxidizing mic-roorganisms. Applied Microbiology and Biotechno-logy, 1998, 50(5): 589–596
Relative Abundance of Anammox Bacterial Marker Genes in the Lower Yangtze River and Their Affecting Factors
Abstract In order to investigate the distribution of anammox bacteria in river ecosystems, water and sediment samples were collected at 6 sites along the lower Yangtze River in spring and autumn in 2014. Clean reads of metagenomic sequencing were BLAST against self-constructed datasets of anammox bacterial marker genes hzsA, hzsB, hzsC and hdh, and valid hits were used to calculate their relative abundances. As results, average relative abundances of hzsA, hzsB, hzsC and hdh genes in water were extremely low due to the excess dissolved oxygen which was not suitable for anammox bacteria. Whereas, the average relative abundances in sediments were much higher, i.e., 4.540×10−10, 4.939×10−10, 4.333×10−10 and 2.859×10−10, respectively. Due to the temperature rise, average relative abundances of hzsA, hzsB, hzsC and hdh genes in sediment samples in autumn were higher than those in spring. From the changes alongside the river, average relative abundances of hzsA, hzsB, hzsC and hdh genes at Datong, Nanjing and Xuliujing were relatively higher, which was due to anthropogenic activity and salinity increase in the estuary. Based on the results of taxonomic identification, anammox bacteria in the lower Yangtze River mainly consisted of Candidatus Brocadia and Candidatus Jettenia at genus level. Relative abundances of hzsA, hzsB, hzsC and hdh genes were related to NO2−-N, NO3−-N and NH4+-N concentration, of which NO2−-N concentration was the restrictive factor as it was much lower than NH4+-N concentration.
Key words lower Yangtze River; anammox bacteria; marker genes; taxonomic composition
doi: 10.13209/j.0479-8023.2019.021
收稿日期: 2018-04-19;
修回日期: 2018-05-16;
网络出版日期: 2019-05-10
国家重点研发计划(2016YFC0402102)资助