基于萃取和超高效液相色谱–串联质谱法的贝类抗生素快速测定方法
徐亚茹 李杰 王之芬 高月 许楠†
深圳市重金属污染控制与资源化重点实验室, 北京大学深圳研究生院环境与能源学院, 深圳 518055; †通信作者, E-mail: xunan@pkusz.edu.cn
摘要建立一种简单、快速的超声波萃取–固相萃取净化–超高效液相色谱串联质谱分析方法, 可以同时测定淡水贝类软组织中磺胺类、喹诺酮类、大环内酯类、β-内酰胺类和氨基糖苷类中15种抗生素。采取超声波萃取法, 对贝类体内的抗生素进行萃取, 用固相萃取方法净化提取液, 最后用超高效液相色谱–三重四级杆质谱对目标物进行分析检测。主要比较两种固相萃取柱Oasis HLB和Oasis PRiME HLB对污染物的净化效率, 后者展现出较好的结果。15种抗生素加标回收率实验结果表明: 当加标浓度为50ng/g时, 回收率为64%~121%, 相对标准偏差为0.5%~19% (n=3); 添加高浓度(100 ng/g)样品时, 回收率为67%~117%, 相对标准偏差为1%~9% (n=3)。方法检测限(LOD)为0.004~0.5 ng/g (干重), 定量限(LOQ)为0.013~1.67 ng/g (干重)。该方法具有回收率高、检测限低的特点。使用该方法对鄱阳湖3个采样点三角帆蚌中的抗生素进行分析, 结果显示, 在15种抗生素中, 有9种均不同程度地检出, 检出频率和浓度最高的是甲氧苄胺嘧啶, 最高浓度达到78.8ng/g, 其次是奇霉素(41.2 ng/g)和环丙沙星(39.8 ng/g)。
关键词抗生素; 贝类; 超声波萃取; 固相萃取; 超高效液相色谱–三重四级杆质谱; Oasis PRiME HLB
抗生素类药物广泛应用于治疗人类和动物的细菌性感染, 大多数药物在生物体内的作用完成后, 80%左右以药物原形排出体外, 不能被生物体完全吸收, 最终通过尿液或者粪便排泄进入环境中[1–2]。此外, 抗生素还可以通过医疗废水和制药废水排入水体。目前已有报道, 各类水体中均检测出不同种类、不同浓度的抗生素[3]。长期的抗生素暴露会对水生生物以及人类产生潜在的毒性效应[4], 特别是废水中富含的抗生素、耐抗生素细菌和抗生素抗性基因, 可以促进生物对抗生素的耐药性[5]。
贝类是一种良好的水环境系统生物指示物[6]。已经有研究表明, 在贝类体内检测到抗生素[7–8]。McEneff等[9]在贻贝中检测到甲氧苄胺嘧啶的浓度是9.22ng/g。Li等[10]在渤海湾的贝类中检测到喹诺酮类、磺胺类和大环内酯类等抗生素, 其中喹诺酮类物质是主要污染物, 最高浓度达到1.575 ng/g。作为复杂的生物基质, 分析检测贝类体内的痕量有机污染物较为困难。
生物样品中抗生素的分析主要包括提取、净化和检测3个主要步骤。微波萃取[11]、加速溶剂萃取[12]、索氏提取[13]以及超声波萃取[14]是使用较多的萃取方法。其中, 超声波萃取因操作简便、节省时间和使用有机溶剂而被广泛应用[15]。萃取后的溶液中含较多杂质, 因此在检测前需要对萃取液进行净化。样品净化常用的方法有液液萃取、微固相萃取和固相萃取(SPE), 其中固相萃取是应用较多的方法[16]。常用的固相萃取柱有Oasis HLB, Oasis WAX, C18和强阴离子交换萃取柱等[17–19]。Oasis PRiME HLB是Waters公司推出的新一代固相萃取产品。与富集性能的Oasis HLB柱子不同, Oasis PRiME HLB是净化性能的固相萃取柱, 无需活化, 高有机相的萃取液可以直接通过柱子, 同时杂质会被柱填料吸收, 目标物直接通过柱子流出, 因此能更快速地处理样品, 并能显著地提高样品洁净度。目前, Oasis PRiME HLB已用于血浆、肾脏和肌肉等样品的分析[20]。
本研究以贝类样品为基质, 超声波萃取抗生素, 固相萃取净化, 并比较固相萃取过程中两种不同萃取柱的净化效果, 使用超高效液相色谱–三重四级杆质谱(UPLC-MS/MS), 建立快速、准确地同时检测贝类样品中多种抗生素的方法, 并考察其应用于实际贝类样品的效果。
1 实验部分
本研究共检测15种不同类别的抗生素, 分别是磺胺嘧啶、磺胺甲基嘧啶、磺胺二甲嘧啶、磺胺甲恶唑、甲氧苄胺嘧啶、磺胺噻唑、磺胺吡啶、环丙沙星、诺氟沙星、氧氟沙星、氧四环素、青霉素G、头孢噻肟钠、奇霉素和克拉霉素。待测抗生素的基本理化性质列于表1。所有抗生素标准品的纯度都高于99%, 均购自德国Dr. Ehrenstorfer公司; 回收率指示物包括购自英国LGC公司的sulfametho-xazole-d4和购于美国TRC公司的roxithromycin-d7。色谱纯甲醇、乙腈购自百灵威公司(J&K, 中国); Oasis HLB和Oasis PRiME HLB (6 mL, 200 mg)购自美国Waters公司; 药用一次性注射器 2.5 mL 购自江西洪达医疗器械集团有限公司; 0.22 μm有机尼龙滤头购自上海安谱实验科技股份有限公司。
用甲醇配置浓度为2mg/L的15种抗生素的混合标准品溶液及浓度为1mg/L的混合内标溶液, 保存在−20°C冰箱中待用。回收率实验的加标浓度是50和100ng/g。对用于定量的标准曲线设置8个浓度梯度, 分别为1, 2, 5, 10, 20, 50, 100和500 ng/g。
2 样品采集与准备
2.1 采样点设置和样品采集
鄱阳湖地处江西省北部, 长江中下游南岸(28°22′—29°45′N, 115°47′—116°45′E), 以松门山为界, 分为南北两部分。北面为入江水道, 长40 km, 宽 3~5 km, 最窄处约2.8 km; 南面为主湖体, 长133 km, 最宽处达74 km。鄱阳湖是中国第一大淡水湖, 也是中国第二大湖。2014年9月, 根据采样点的水文条件和三角帆蚌的生活习性, 在鄱阳湖设置3个不同的采样点(图1), 分别是修水、赣江和鄱阳。使用蚌耙和钩子采集三角帆蚌样品, 冷藏运回实验室, 测定贝类的长度和重量, 然后放于装水的干净容器中24小时, 以除去一些杂质。分离贝类的软组织, 冷冻干燥处理后, 用锡纸包好后, 置于−20ºC冰箱中备用。
表1 目标抗生素的基本理化性质
Table 1 Target antibiotics and their physicochemical properties
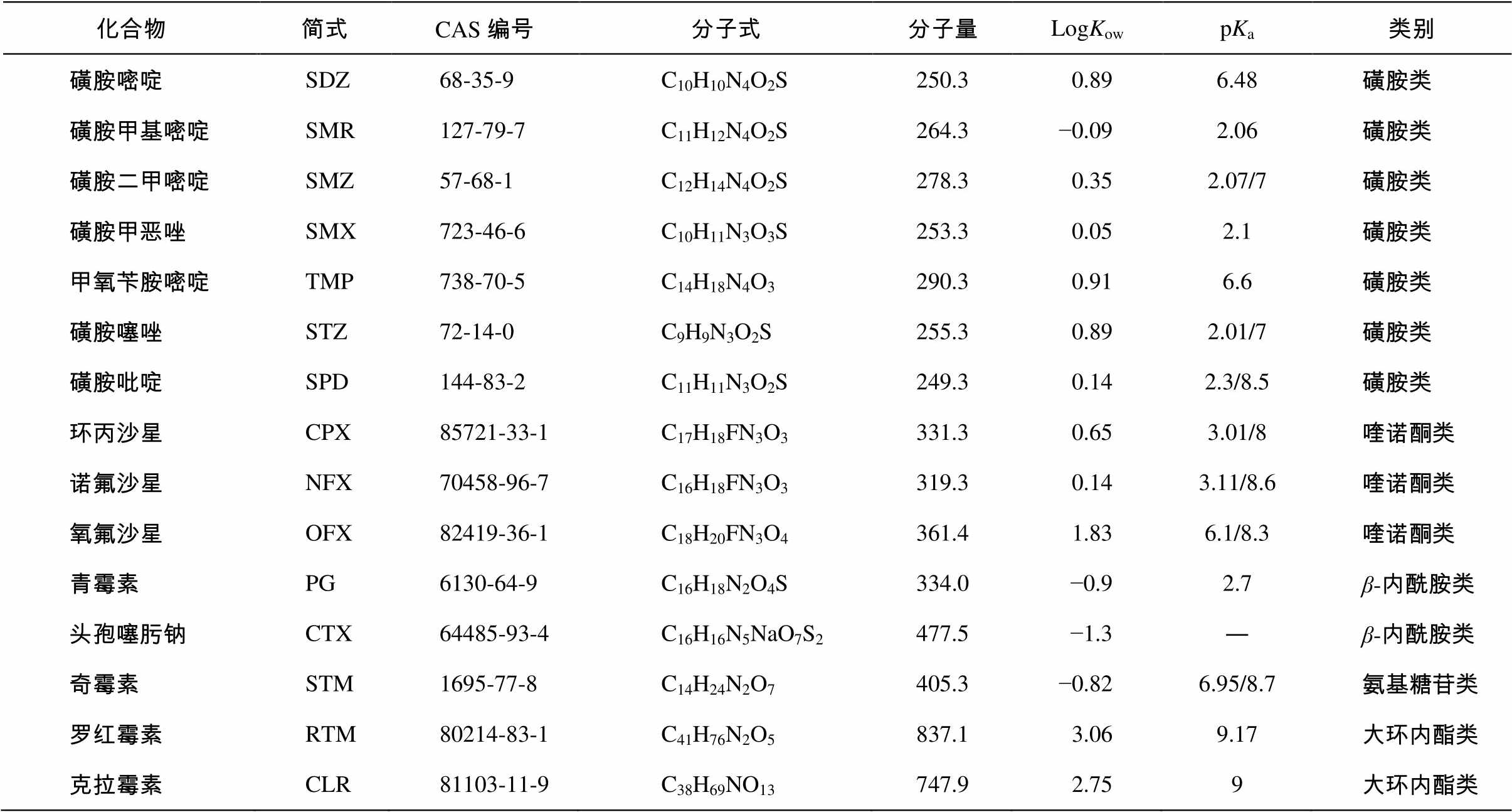
化合物简式CAS编号分子式分子量LogKowpKa类别 磺胺嘧啶SDZ68-35-9C10H10N4O2S250.30.896.48磺胺类 磺胺甲基嘧啶SMR127-79-7C11H12N4O2S264.3−0.092.06磺胺类 磺胺二甲嘧啶SMZ57-68-1C12H14N4O2S278.30.352.07/7磺胺类 磺胺甲恶唑SMX723-46-6C10H11N3O3S253.30.052.1磺胺类 甲氧苄胺嘧啶TMP738-70-5C14H18N4O3290.30.916.6磺胺类 磺胺噻唑STZ72-14-0C9H9N3O2S255.30.892.01/7磺胺类 磺胺吡啶SPD144-83-2C11H11N3O2S249.30.142.3/8.5磺胺类 环丙沙星CPX85721-33-1C17H18FN3O3331.30.653.01/8喹诺酮类 诺氟沙星NFX70458-96-7C16H18FN3O3319.30.143.11/8.6喹诺酮类 氧氟沙星OFX82419-36-1C18H20FN3O4361.41.836.1/8.3喹诺酮类 青霉素PG6130-64-9C16H18N2O4S334.0−0.92.7β-内酰胺类 头孢噻肟钠CTX64485-93-4C16H16N5NaO7S2477.5−1.3―β-内酰胺类 奇霉素STM1695-77-8C14H24N2O7405.3−0.826.95/8.7氨基糖苷类 罗红霉素RTM80214-83-1C41H76N2O5837.13.069.17大环内酯类 克拉霉素CLR81103-11-9C38H69NO13747.92.759大环内酯类
说明: 数据来源于物竞化学品数据库(http://www.basechem.org)。

图1 鄱阳湖采样点分布
Fig. 1 Location map of sampling sites in Poyang Lake
2.2 淡水贝类样品前处理
2.2.1 超声波萃取
用研钵将干燥后的样品研磨成粉末状。称取0.2±0.01g粉末状样品, 置于50mL的离心管中, 加入上述混合内标液100μL, 混匀, 置于通风橱中过夜。取8mL萃取剂加入离心管中, 涡旋1分钟使其充分混合, 然后在42kHz条件下超声波萃取15分钟, 再5000rpm离心5分钟, 转移上清液至干净的离心管。重复上述萃取操作1次, 合并两次萃取所得的上清液约16 mL, 待SPE净化。
2.2.2 固相萃取
采用24孔的固相萃取装置(Supelco, Bellefonte, PA, 美国)进行固相萃取。本研究比较Oasis PRiME HLB在不同萃取剂条件下以及Oasis HLB在不同pH条件下的萃取效率。
Oasis PRiME HLB的萃取条件主要参考Waters公司检测猪肉中农兽药残留的方法[21]。超声波萃取剂分别采用80%乙腈和80%甲醇, 对比二者的萃取效果。取10mL萃取液, 直接通过OasisPRiMEHLB固相萃取小柱净化。该萃取柱是净化柱, 柱子的填料吸附污染物, 目标物会直接流出。收集流出的液体于10mL的玻璃小管中, 35ºC氮气吹干至0.1mL, 用UPLC-MS/MS初始流动相(乙腈:水=1:1, 体积比)定容至1mL, 用0.22μm的针式过滤器过滤后, 转移至玻璃进样小瓶, 待测。
Oasis HLB萃取条件参考美国EPA 1694方法[22], 选取乙腈作为超声波萃取剂进行回收率实验, 采用Oasis HLB小柱进行固相萃取。本研究分析的物质理化性质差异较大, 通过一次萃取全部检出分析物比较困难, 所以首先需要调节萃取液的pH值。实验中对比未调节萃取液pH值(接近中性)及调节pH值(分别为2和10)3种条件。随后, 将萃取液溶解在400mL蒸馏水中, 以降低萃取液中有机相的比例。在溶液中加入0.2gNa4EDTA, 可以与样品中的金属离子络合, 减少对抗生素分析结果的影响。Oasis HLB属于富集柱, 待测目标物会被柱填料吸附。使用OasisHLB前, 分别用5mL甲醇和5mL蒸馏水对柱子进行活化, 待萃取液全部通过固相萃取柱后, 用10mL超纯水淋洗柱子, 真空抽干约1小时, 最后用10mL甲醇洗脱目标物。洗脱液收集在10mL的玻璃小管中, 在35ºC下经氮气吹干至0.1mL, 用UPLC- MS/MS初始流动相(乙腈:水=1:1, 体积比)定容至1mL, 用0.22μm的针式过滤器过滤后, 转移至玻璃进样小瓶, 待测。
2.3 液相色谱和质谱条件
采用UPLC-MS/MS(Liquid Chromatography 1290 series RRLC system和6460 triple quadrupole MS, Agilent, 美国)对目标分析物进行检测, XDB-C18分析柱(4.6 mm × 50 mm, 1.8 μm 孔径, Agilent, 美国)。流动相A为0.1%(体积分数)甲酸/水溶液, 流动相B是100%的乙腈, 流速为0.3mL/min。梯度洗脱程序: 初始浓度梯度是10% B 保持1分钟, 流速为0.3mL/min, 在20.1分钟内, 浓度由10%B增加至90% B, 保持0.1分钟, 然后在1分钟后回到初始浓度10%B, 最后平衡5分钟, 进样体积为2μL, 柱温保持在25ºC。
质谱参数: 电喷雾电离源(ESI), 毛细管电压为+4000V, 雾化气压力为50psi, 气体流速为10L/ min, 干燥气温度为350ºC。所有分析物的定量采用多反应监测(MRM)正离子模式, 检测得到的数据使用Masshunter软件分析。目标抗生素的色谱检测条件如表2所示。
3 结果与讨论
3.1 固相萃取条件的选择与优化
除萃取柱的类型和化合物的性质外, 洗脱剂用量及组成、上样流速、样品pH等因素均会影响固相萃取的回收率。本研究比较萃取柱Oasis HLB在3种不同pH条件下的表现和Oasis PRiME HLB在两种不同萃取剂条件下的表现, 最后综合比较两种萃取柱的萃取效果。
表2 目标抗生素的色谱检测条件及方法检测限和定量限
Table 2 Chromatographic conditions, LOD and LOQ of target antibiotics

化合物tR/min定量离子m/z定性离子m/zLOD/ (ng·g−1)LOQ/ (ng·g−1) SDZ3.3171562510.0540.180 SPD4.0331562500.0050.019 STZ4.1491562560.0090.030 SMZ5.6251562790.0070.024 SMR4.4991562650.0040.013 SMX9.8321562540.0130.040 TMP4.4642302910.0070.024 OFX6.0233183620.0140.049 NFX6.0772442620.5001.666 CPX6.5463143320.1360.454 CLR16.2791587480.0740.249 RTM16.8701588380.0270.091 PG13.6332173350.0400.133 CTX5.7693964560.0880.294 STM6.5452893330.1670.555
3.1.1 Oasis HLB的萃取效果
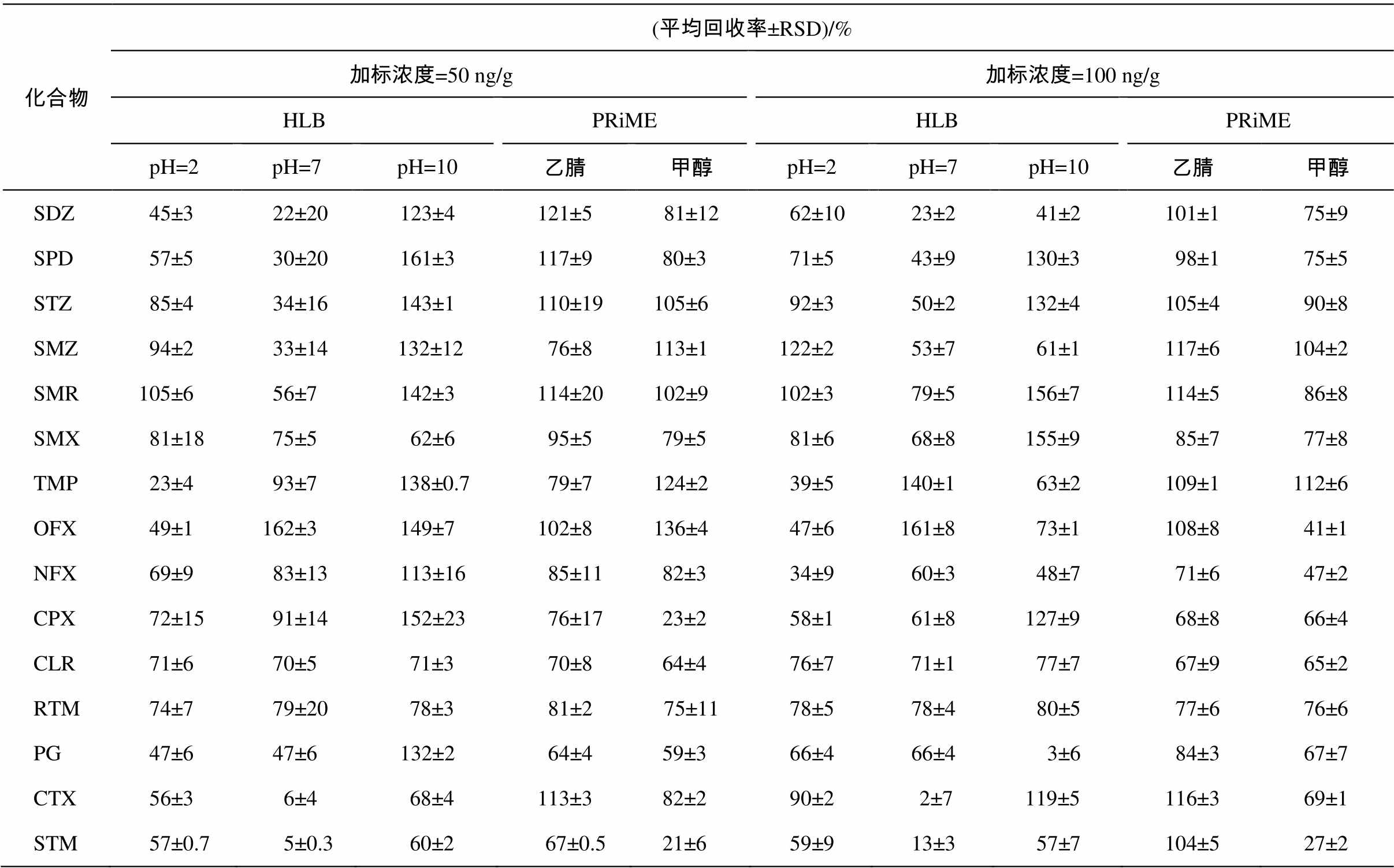
表3列出两种固相萃取小柱在不同萃取条件下对贝类样品中抗生素的回收率。结果表明, pH调节为2时, 大多数目标物的回收率在48%~118%之间, 只有甲氧苄胺嘧啶的回收率低于25%。不调节pH时, 大多数目标物的回收率范围是22%~162%, 环丙沙星和奇霉素的回收率低于10%, 而氧氟沙星的回收率大于120%。pH调节为10时, 待测物的回收率高于60%, 但大多数物质的回收率高于120%。由此可见, 极性物质的回收率受pH的影响较大, 对酸性条件敏感的物质(如磺胺二甲嘧啶)在酸性条件下回收率较高, 对碱性条件敏感的物质(如甲氧苄胺嘧啶)在碱性条件下回收率比较好。此外, 贝类样品一般包含15%的脂质, 尽管这些脂质不易溶于乙腈, 但还会有一些杂质被萃取出来[23], 从而对回收率产生影响。
3.1.2 Oasis PRiME HLB的萃取效果
使用Oasis PRiME HLB作为固相萃取柱时, 比较80%乙腈溶液和80%甲醇溶液作为萃取剂的萃取效率(表3)。80%甲醇溶液作为萃取剂时, 目标物的回收率范围是20%~130%, 其中环丙沙星和奇霉素的回收率均低于40%。80%乙腈溶液作为萃取剂时, 目标物的回收率范围是64%~121%, 表明待测物更倾向溶于乙腈中。
表3 加标浓度为50 ng/g和100 ng/g时不同净化条件下贝类样品中抗生素的平均回收率(±相对标准偏差)(n=3)
Table 3 Average recoveries (±relative standard deviation) of antibiotics in mussels using different clean-up approaches spiked at 50 ng/g and 100 ng/g (n=3)
化合物(平均回收率±RSD)/% 加标浓度=50 ng/g加标浓度=100 ng/g HLBPRiMEHLBPRiME pH=2pH=7pH=10乙腈甲醇pH=2pH=7pH=10乙腈甲醇 SDZ45±322±20123±4121±581±12 62±1023±241±2101±175±9 SPD57±530±20161±3117±980±371±543±9130±3 98±175±5 STZ85±434±16143±1 110±19105±692±350±2132±4105±490±8 SMZ94±233±14132±12 76±8113±1122±253±761±1117±6104±2 SMR105±656±7142±3 114±20102±9102±379±5156±7114±586±8 SMX81±1875±5 62±6 95±579±581±668±8155±9 85±777±8 TMP23±493±7 138±0.7 79±7124±239±5140±163±2109±1112±6 OFX49±1162±3149±7102±8136±447±6161±873±1108±841±1 NFX69±983±13 113±16 85±1182±334±960±348±7 71±647±2 CPX72±1591±14 152±23 76±1723±258±161±8127±9 68±866±4 CLR71±670±571±3 70±864±476±771±177±7 67±965±2 RTM74±779±2078±3 81±275±1178±578±480±5 77±676±6 PG47±647±6132±2 64±459±366±466±4 3±6 84±367±7 CTX56±36±468±4113±382±290±22±7119±5116±369±1 STM57±0.75±0.360±2 67±0.521±659±913±357±7104±527±2
只有在使用Oasis PRiME HLB时, 奇霉素、克拉霉素和罗红霉素才能获得较好的回收率, 说明使用Oasis PRiME HLB时, 能在较短时间内获得更纯净、种类更多的目标物。
3.2 方法的准确性
3.2.1 回收率
准确称取0.2±0.01g研磨后的贝类样品作为基质, 选取2个加标浓度级别, 分别为50和100ng/g。利用所建立的超声波萃取–OasisPRiMEHLB固相萃取净化–超高效液相色谱–串联质谱分析法对样品进行检测, 每个浓度水平有3个重复, 用来考察方法的总体回收率和重现性。每个浓度水平设置两个空白样品, 用来检验实验过程中的干扰。回收率指示物在萃取前加入, 对整个实验过程进行质量控制。15种抗生素的回收率如表3所示。当添加浓度为50ng/g时, 回收率为64%~121%, RSD是0.5%~19% (n=3); 添加浓度为100 ng/g时, 回收率是67%~117%, RSD是1%~9% (n=3)。结果表明该方法具有较好的重现性, 精密度和回收率也符合要求。
3.2.2 线性和检测限
方法的线性通过以下步骤测定: 配制8个不同浓度梯度的标准品溶液, 浓度分别为1, 5, 10, 50, 100, 200和500 mg/L, 经过仪器检测, 测定主峰的面积, 计算响应含量, 以含量为横坐标, 峰面积为纵坐标, 得到线性回归方程 (R2>0.999)。
方法的灵敏度用检测限(LOD)和定量限(LOQ)进行表征。LOD是信噪比为3时的样品浓度, 变化范围是0.004~0.5ng/g; LOQ是信噪比为10时的样品浓度, 变化范围是 0.013~1.67ng/g。结果表明, 建立的方法灵敏度高(表 2)。
3.3 实际样品分析
为了证明方法的可靠性, 我们运用该方法对鄱阳湖 3 个不同采样点三角帆蚌体内的抗生素进行检测。在 15 种抗生素中, 有 9 种的检出频率为 100%(图 2)。甲氧苄胺嘧啶的检出浓度最高, 最高浓度达到 78.8ng/g, 其次是奇霉素(41.2ng/g)和环丙沙星(39.8 ng/g)。
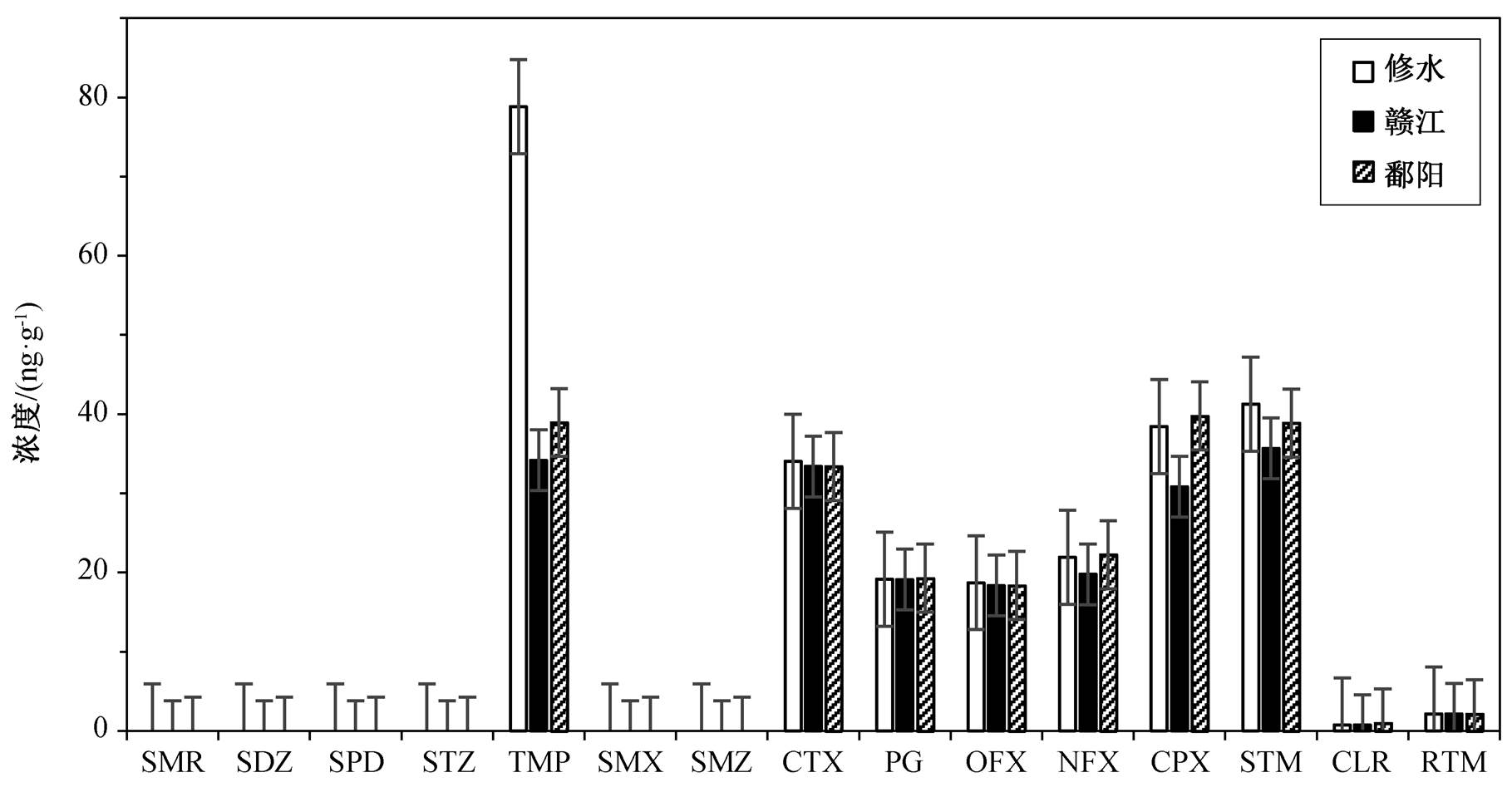
图2 鄱阳湖3个断面的贝类中抗生素的浓度分布
Fig. 2 Concentrations of target antibiotics in mussels from three sampling sites along Poyang Lake
大多数磺胺类物质在三角帆蚌中均未检出, 说明三角帆蚌中磺胺类物质的浓度可能低于检测限。还有一个原因是大多数磺胺类物质易溶于水, 不易在生物体中累积。甲氧苄胺嘧啶也属于磺胺类, 但检出浓度和频率都很高, 这是由于甲氧苄胺嘧啶的pKa为 6.6, 在偏碱性的水环境中不会完全电离[9]。环丙沙星极易吸附于微粒中, 导致其在贝类中的浓度较高[24]。此外, 罗红霉素和克拉霉素的检出浓度比较低, 这是由于它们在贝类体内代谢较快, 不易累积[25]。
除甲氧苄胺嘧啶外, 3 个断面三角帆蚌中检出的抗生素种类和浓度基本上一致, 可见空间的差异对抗生素在贝类体内的分布影响不大。从整体上来说, 与其他文献报道相比, 贝类中抗生素的检出浓度处于中等偏高的水平[26]。
4 小结
本研究基于 EPA1694 方法, 对样品的萃取与净化过程进行改进, 超声波萃取后, 在 SPE 中使用 OasisPRiMEHLB 作为固相萃取小柱, 并联合UPLC–MS/MS 检测, 建立了高效、快速的检测淡水贝类软组织中抗生素含量的方法。通过验证回收率、检测限和定量限, 证明该方法符合检测标准, 并成功应地用于实际贝类样品。结果显示, 三角帆蚌软组织中 15 种抗生素有 9 种均不同程度地检出, 检出频率和浓度最高的是甲氧苄胺嘧啶(78.8ng/g), 其次是奇霉素(41.2ng/g)和环丙沙星(39.8ng/g)。实际样品的检测表明贝类受到不同程度的污染, 这些抗生素的来源以及风险需要进一步研究。贝类组织中抗生素检测分析方法的建立对了解抗生素在生物体内的分布、迁移转化规律和生物毒理性质具有重要意义, 可以为抗生素的生态风险评价及环境生态修复提供科学依据。
参考文献
[1]Loos R, Carvalho R, Antonio D C, et al. EU-wide monitoring survey on emerging polar organic con-taminants in wastewater treatment plant effluents. Water Res, 2013, 47(17): 6475–6487
[2]俞道进, 曾振灵, 陈杖榴. 四环素类抗生素残留对水生态环境影响的研究进展. 中国兽医学报, 2004, 24(5): 515–517
[3]Manzetti S, Ghisi R. The environmental release and fate of antibiotics. Mar Pollut Bull, 2014, 79(1/2): 7–15
[4]Kummerer K. Antibiotics in the aquatic environment — a review — part I. Chemosphere, 2009, 75(4): 417–434
[5]Heuer O E, Kruse H, Grave K, et al. Human health consequences of use of antimicrobial agents in aqua-culture. Clin Infect Dis, 2009, 49(8): 1248–1253
[6]Anderson A S, Chris M W, Adalto B, et al. Responses of biomarkers in wild freshwater mussels chronically exposed to complex contaminant mixtures. Ecotoxico-logy, 2014, 23(7): 1345–1358
[7]Klosterhaus S L, Grace R, Hamilton M C, et al. Method validation and reconnaissance of pharmaceu-ticals, personal care products, and alkylphenols in sur-face waters, sediments, and mussels in an urban estuary. Environ Int, 2013, 54(5): 92–99
[8]Nakata H, Shinohara R, Nakazawa Y, et al. Asia-Pacific mussel watch for emerging pollutants: distri-bution of synthetic musks and benzotriazole UV stabi-lizers in Asian and US coastal waters. Mar Pollut Bull, 2012, 64(10): 2211–2218
[9]McEneff G, Barronc L, Kellehe B, et al. A year-long study of the spatial occurrence and relative distribu-tion of pharmaceutical residues in sewage effluent, receiving marine waters and marine bivalves. Science of the Total Environment, 2014, 476/477: 317–326
[10]Li W, Shi Y, Gao L, et al. Investigation of antibiotics in mollusks from coastal waters in the Bohai Sea of China. Environ Pollut, 2012,162(5): 56–62
[11]Bayen S, Lee H K, Obbard J P. Determination of polybrominated diphenyl ethers in marine biological tissues using microwave-assisted extraction. Journal of Chromatography A, 2004, 1035(2): 291–294
[12]Ramos L. Critical overview of selected contemporary sample preparation techniques. Journal of Chromato-graphy A, 2012, 1221(5): 84–98
[13]Stevens J L, Northcott G L, Stern G A. PAHs, PCBs, PCNs, organochlorine pesticides, synthetic musks, and polychlorinated nalkanes in UK sewage sludge: survey results and implications. Environ Sci Technol, 2013, 37(3): 462–467
[14]Yu Y, Wu L S. Analysis of endocrine disrupting com-pounds, pharmaceuticals and personal care products in sewage sludge by gas chromatography-mass spec-trometry. Talanta, 2012, 89(2): 258–263
[15]Tanoue R, Nomiyama K, Nakamura H, et al. Simultaneous determination of polar pharmaceuticals and personal care products in biological organs and tissues. Journal of Chromatography A, 2014, 1355: 193–205
[16]Carballa M, Omil F, Lema J M, et al. Behavior of pharmaceuticals, cosmetics and hormones in a sewage treatment plant. Water Res, 2004, 38(8): 2918–2926
[17]Unno T, Sagesaka Y M, Kakuda T. Analysis of tea catechins in human plasma by high-performance liquid chromatography with solid-phase extraction. J Agric Food Chem, 2005, 53(2): 9885–9889
[18]Zhou Zhen, Shi Yali, Li Wenhui, et al. Perfluorinated compounds in surface water and organisms from Baiyangdian Lake in North China: source profiles, bioaccumulation and potential risk. Bull Environ Contam Toxicol, 2012, 89(3): 519–524
[19]Elena M, Meritxell G, Sílvia L, et al. Simplified procedures for the analysis of polycyclic aromatic hydrocarbons in water, sediments and mussels. Jour-nal of Chromatography A, 2004, 1047(2): 181–188
[20]Pisarcikova J, Ocelova V, Faix S, et al. Identification and quantification of thymol metabolites in plasma, liver and duodenal wall of broiler chickens using UHPLC-ESI-QTOF-MS. Biomedical Chromatography, 2016, 31(5): 1–12
[21]黄德凤, Tran K V, Young M S, 等. Oasis PRiME HLB-UPLC-MS/MS测定猪肉中78种兽药残留[EB/ OL]. (2015-07) [2017-04-01]. http://www.waters.com/webassets/cms/library/docs/china_Oasis_PRiME_HLB_determine_78_residues_in_pork.pdf
[22]US Environmental Protection Agency. Method 1694: pharmaceuticals and personal care products in water, soil, sediment, and biosolids by HPLC/MS/MS, EPA-821-R-08-002 [R/OL]. (2007–12) [2017-04-01]. https://www.epa.gov/sites/production/files/2015-10/docu ments/method_1694_2007.pdf
[23]Liu A, Tweed J, Wujcik C E. Investigation of an on-line two-dimensional chromatographic approach for peptide analysis in plasma by LC-MS-MS. J Chroma-togr B: Analyt Technol Biomed Life Sci. 2009, 877 (20/21): 1873–1881
[24]Gao L, Shi Y, Li W, et al. Occurrence, distribution and bioaccumula-tion of antibiotics in the Haihe River in China. J Environ Monit, 2012, 14(3): 1248–1255
[25]Peng X, Zhang K, Tang C, et al. Distribution pattern, behavior, and fate of antibacterials in urban aquatic environments in South China. J Environ Monit, 2011, 13(2): 446–454
[26]Santos L, Soares1 B, Rosa J, et al. Detection and quantification of 41 antibiotic residues in gilthead sea bream (Sparus aurata) from aquaculture origin, using a multiclass and multi-residue UPLC-MS/MS method. Food Anal Methods, 2016, 9(10): 2749–2753
Fast Determination of Multiple Antibiotics in Mussels Based on Extraction and UPLC-MS/MS
XU Yaru, LI Jie, WANG Zhifen, GAO Yue, XU Nan†
Key Laboratory for Heavy Metal Pollution Control and Reutilization, School of Environment and Energy, Peking University Shenzhen Graduate School, Shenzhen 518055; † Corresponding author, E-mail: xunan@pkusz.edu.cn
AbstractA fast and simple method for simultaneous determination of fifteen antibiotics (sulfadiazine, ciprofloxa- cin, penicillin, spechromycin and roxithromycin) in mussels using ultrasound extraction, solid phase extraction purification and ultra performance liquid chromatography-tandem mass spectrometric was established. The mussel samples were extracted by ultrasonic extraction, followed by solid-phase extraction (SPE) as the cleanup proce-dure. Ultra performance liquid chromatography-triple quadrupole mass spectrometry (UPLC-MS/MS) was used for identification and quantification of antibiotics. The performance of two kinds of SPE cartridges, Oasis HLB and Oasis PRiME HLB, was compared, with the latter showing better results. Recoveries ranged from 64% to 121% at spiking levels of 50 ng/g, with RSD being 0.5%–19%, and from 67% to 117% at spiking levels of 100 ng/g, with RSD being 1%–9%. Limits of detection (LODs) ranged from 0.004 to 0.5 ng/g (dry weight) and limits of quantifi-cation (LOQs) ranged from 0.013 to 1.67 ng/g (dry weight). The recoveries were reasonable and the detection limit was low. Finally, the method was successfully applied to the determination of the target antibiotics in mussel samples (Hyriopsis cumingii) collected from Poyang Lake. 9 out of 15 selected antibiotics were detected in the collected mussel tissues. The concentration and detection frequency of trimethoprim in mussels were the highest. The concentration of trimethoprim was 78.8 ng/g, followed by spectinmycin (41.2 ng/g) and ciprofloxacin (39.8 ng/g).
Key wordsantibiotics; mussels; ultrasonication extraction; solid-phase extraction; UPLC-MS/MS; Oasis PRiME HLB
中图分类号X703
doi:10.13209/j.0479-8023.2018.009
国家自然科学基金(51579003)和深圳市科技计划(JCYJ20150629144818001)资助
收稿日期:2017-05-17;
修回日期:2017-05-24;
网络出版日期:2018-04-03