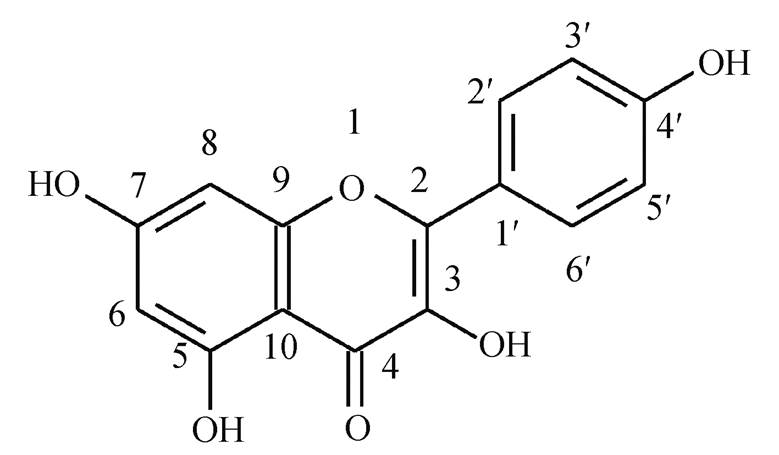
图1 山奈酚的分子结构及原子编号
Fig. 1 Molecular structure and atomic numbering of kaempferol
韩旭泽 晏明全 †
水沙科学教育部重点实验室, 北京大学环境工程系, 北京 100871
摘要 为了揭示山奈酚的解离及金属络合过程机理, 测定山奈酚在解离(pH=2.01~13.00)和与铜离子络合(0.1~2710.9μmol/L)过程的紫外–可见光光谱, 经过差分处理得到各反应阶段的特征光谱。结合量子化学理论计算方法, 基于密度泛函理论以及含时密度泛函理论, 计算可能的各种去质子化、络合结构及对应的电子吸收光谱。通过对比实验值与计算值发现: 解离过程按7, 4′, 3, 5号酚基的顺序依次去质子化; 在低浓度Cu 2+ (<67.9 μmol/L)时, 4号和5号酚基与Cu 2+ 络合; 在高浓度Cu 2+ (67.9~2710.9 μmol/L)时, 3号和4号酚基与Cu 2+ 络合。在山奈酚的解离、与Cu 2+ 络合过程中, 计算光谱与差分光谱能够很好地吻合, 特征峰的出现是由于解离、络合前后活性基团电子分布的变化、光子照射时最高占据分子轨道到最低未占据分子轨道能量吸收的变化导致,揭示了山奈酚解离及金属络合过程的机理。
关键词 差分光谱; 量子化学计算; 解离; 络合; 分子轨道
山奈酚是一种天然黄酮类, 是许多植物性食物和中药中存在的次级代谢产物, 尤其在蕨类植物和被子植物中较为常见 [1] 。人类的正常饮食中, 每天平均摄入23mg黄酮类化合物, 其中约17%为山奈酚 [2] 。常见的富含山奈酚的蔬菜和水果包括土豆、洋葱、西兰花、南瓜、黄瓜、莴苣、菠菜、苹果、葡萄、桃子和黑莓 [3] 。山奈酚能够减少氧化应激, 因此可作为抗氧化剂 [4] 。研究表明, 山奈酚具有多种生物学作用, 能够降低各种癌症风险, 是潜在的癌症治疗手段 [3] 。
山奈酚属于多酚化合物, 是有效的天然金属螯合有机物, 影响环境中金属离子的赋存形态和迁移转化, 环境行为效应较复杂 [4] 。山奈酚具有4个酚基(分子结构见图1), 意味着随着环境中pH的变化, 山奈酚的酸碱形式将发生改变。已有研究表明, pH影响山奈酚的抗氧化活性 [1] 。当山奈酚解离后, 酸碱形式的变化将导致其与金属离子的络合结构发生变化, 从而影响其化学性质。这种变化能够反映在紫外–可见光(UV-vis)光谱上。因此, 掌握山奈酚的解离及金属络合机理, 是研究其药物治疗效果和环境影响效应的基础。
本文以山奈酚为研究对象, 测定其在解离(pH2.01~13.00)过程和与铜离子(0.1~2710.9 μmol/L)络合过程中的紫外–可见光光谱, 经过差分处理可得到各反应阶段的特征光谱, 结合量子化学理论计算方法, 基于密度泛函理论(density functionaltheory, DFT)和含时密度泛函理论(time-dependent density functional theory, TD-DFT), 计算可能的各种去质子化、络合结构及对应的电子吸收光谱。通过实验值与计算值的比较, 研究其微观反应过程, 揭示其解离及金属络合过程的机理。
图1 山奈酚的分子结构及原子编号
Fig. 1 Molecular structure and atomic numbering of kaempferol
山奈酚C 15 H 10 O 6 购自Aladdin公司, 高氯酸铜Cu(ClO 4 ) 2 和高氯酸钠NaClO 4 购自Sigma公司, 高氯酸HClO 4 购自国药集团, 氢氧化钠NaOH购自北京化工厂。为减少离子对吸收光谱的干扰, 选用高氯酸铜作为金属离子来源。高氯酸钠作为背景电解质, 用于保持离子强度恒定。
UV-vis分析使用U-3900型紫外–可见光光谱仪(日本HITACHI公司), 采用5cm比色皿, 扫描波长为200~600nm, 间隔1nm, 基线误差为±0.0005 cm −1 。解离过程的差分光谱通过各阶段初始pH时的吸收光谱作差处理得到, 络合过程的差分光谱通过各阶段初始Cu 2+ 浓度时的吸收光谱作差处理得到。
1)解离过程。配制山奈酚溶液1.5mg/L于100 mL锥形瓶中, 投入搅拌子, 置于磁力搅拌器合适转速下, 将初始pH调至2.0(±0.03), 稳定30分钟。逐步滴加NaOH, 以0.33为增量将pH调至13.0, 每次等待稳定时间10分钟, 取样测其UV-vis光谱, 取样期间须保证pH稳定 [5] 。
2)金属络合过程。配制山奈酚溶液1.5mg/L于100mL锥形瓶中, 加入1mL 1mol/L NaClO 4 溶液, 控制离子强度为0.01mol/L。投入搅拌子, 置于磁力搅拌器合适转速下, 将初始pH调至5.0(±0.03), 稳定30分钟。逐步滴加0.001, 0.01, 0.1和1mol/L Cu(ClO 4 ) 2 溶液, 使山奈酚溶液中Cu 2+ 浓度从0增至2710.9μmol/L, 每次等待稳定时间10分钟, 取样测其UV-vis光谱, 取样期间须保证pH稳定 [6] 。
本研究中相关计算均使用Gaussian 09软件 [7] 实现, 主要原理基于密度泛函理论(DFT)及其衍生的含时密度泛函理论(TD-DFT) [8] 。计算方法为B3LYP杂化泛函 [9–10] , 使用DFT方法优化结构, 对H, C, O和Cu原子均使用6-31g(d,p)基组, 结构优化不设置对称性约束; 使用TD-DFT方法计算紫外–可见光光谱, 对各原子均使用6-311+g(d,p)基组 [11] 。
山奈酚酸碱滴定实验光谱见图 2, 可以看到滴定过程中光谱信号变化复杂, 根据pH增加过程能够逐步发现光谱的依次演化方向。
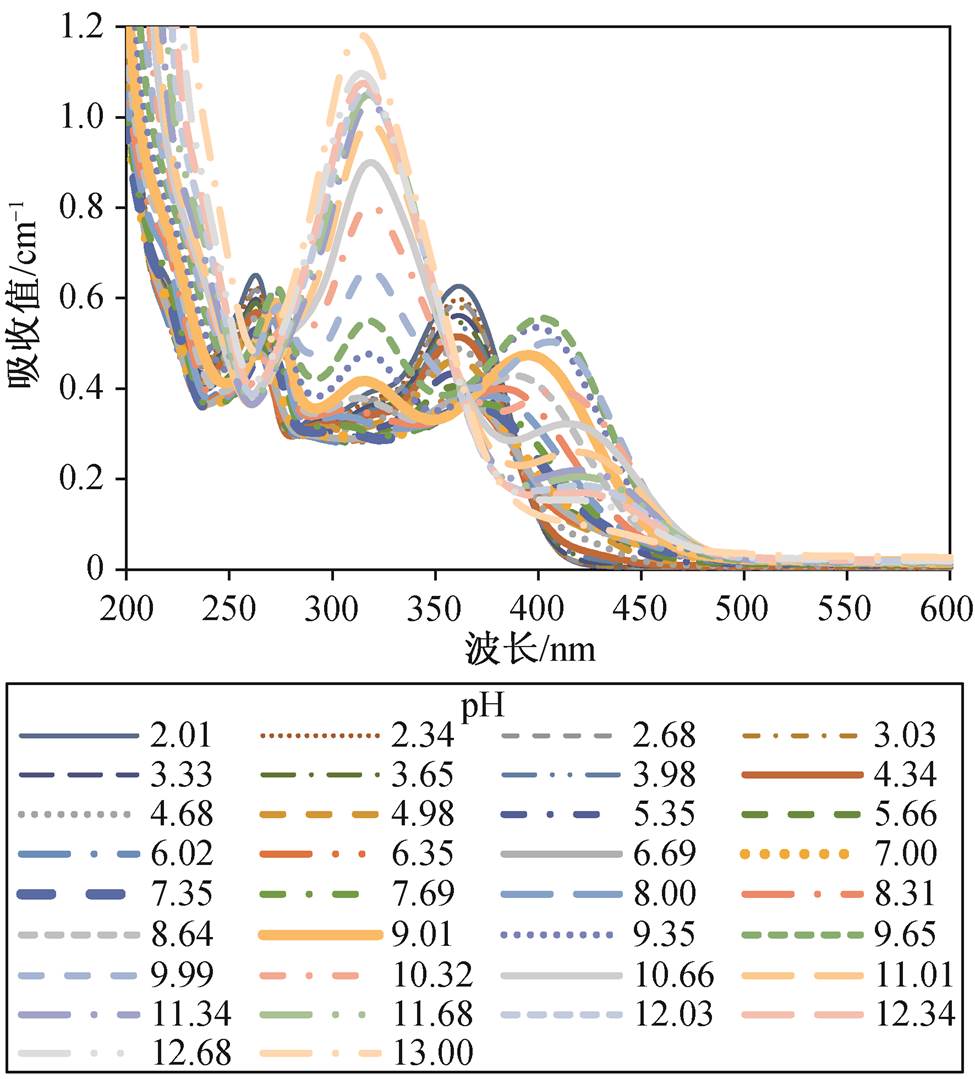
图2 不同pH值下山奈酚的吸收光谱
Fig. 2 Absorbance spectra of kaempferol at various pH values
通过作差处理, 发现解离过程的差分光谱随pH增大具有阶段性规律, 每个阶段具有各自的光谱变化特征, 这与其分子结构密切相关。从其分子结构(图1)可知, 山奈酚的3号、5号、7号和4′号位均为酚基。随着pH增高, 4个酚基相继发生解离(去质子化)作用, 每一次去质子化导致分子结构和电子分布的改变, 从而引起UV-vis光谱发生变化。因此, 山奈酚各阶段去质子化位点的不同引起光谱之间的差异是导致差分光谱具有阶段性的决定因素, 这可以通过量子化学计算各去质子化结构的UV-vis吸收光谱得到进一步证实。
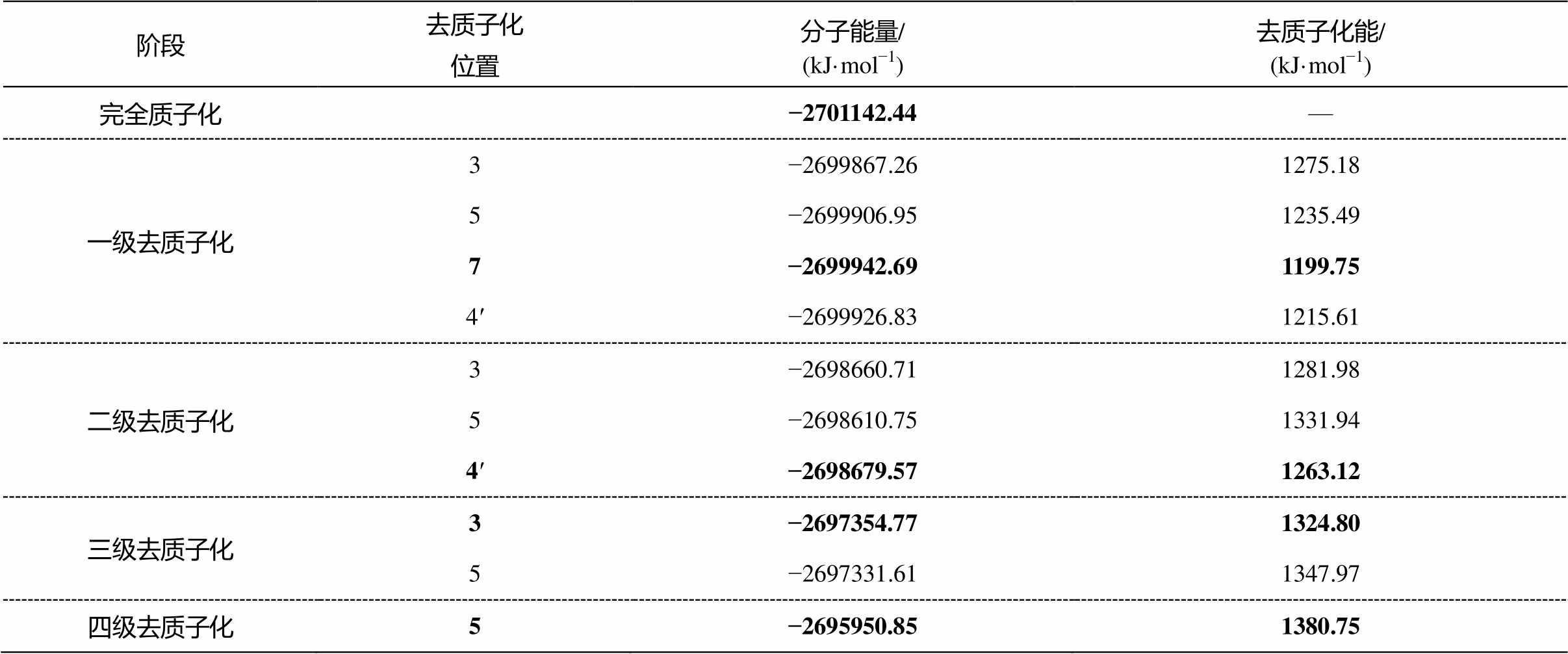
分子具有由原子核和电子运动产生的动能及其相互吸引、排斥产生的势能, 总能量为动能与势能之和。由能量最低原理可知, 化学反应总是向着具有最低能量差的方向进行。因此, 若每一次去质子化时所有可能的反应能量差为已知, 即可通过判断能量差大小得到反应顺序。通过计算山奈酚每一步去质子化时可能去质子化结构的Gibbs自由能(表1), 比较其与上一步得到的去质子化结构(表1中加粗数字)的能量差, 具最小能量差的结构即为本步骤的去质子化结构, 依次计算一、二、三、四级去质子化过程, 得到去质子化顺序。
通过上述能量计算, 可获得山奈酚在水溶液中随pH增大的去质子化顺序为完全质子化(未去质子化)、7号、4′号、3号和5号。在此基础上, 依次计算完全质子化(fully pro)、7号去质子化(7-de)、4′号去质子化(7-4′-de)、3号去质子化(7-4′-3-de)以及5号去质子化(7-4′-3-5-de)共5种去质子化结构的紫外–可见光光谱(图3), 并逐级作差, 与对应的4个阶段实验差分光谱进行对比(图 4)。
表 1 每一步去质子化时所有可能去质子化结构的 Gibbs自由能比较
Table 1 Comparison of Gibbs free energies of each possible deprotonated structure for successive deprotonation
在pH2.01增至5.66过程中, 山奈酚的差分实验光谱显示在211, 233, 252, 277, 328和414 nm处出峰, 计算差分光谱中出现的230和278 nm分别与实验值非常接近, 316和393 nm分别与实验值较为接近, 均在TD-DFT计算的典型误差范围(40nm波长, 或0.2eV激发能量)内 [12–13] 。在pH5.66增至9.65过程中, 山奈酚的差分实验光谱显示在219, 274, 315和407 nm处出峰, 计算差分光谱中出现的212 nm与实验值非常接近, 275和315nm与实验值几乎完全一致, 484 nm与实验值差距较大。在pH 9.65增至11.34过程中, 山奈酚的差分实验光谱显示在237, 318和465 nm处出峰, 计算差分光谱中出现的237nm与实验值完全一致, 314nm与实验值非常接近, 525nm与实验值差距较大。在pH11.34增至13.00过程中, 山奈酚的差分实验光谱显示在223, 303和351nm处出峰, 计算差分光谱中出现的228和293nm分别与实验值非常接近, 413 nm与实验值差距较大。
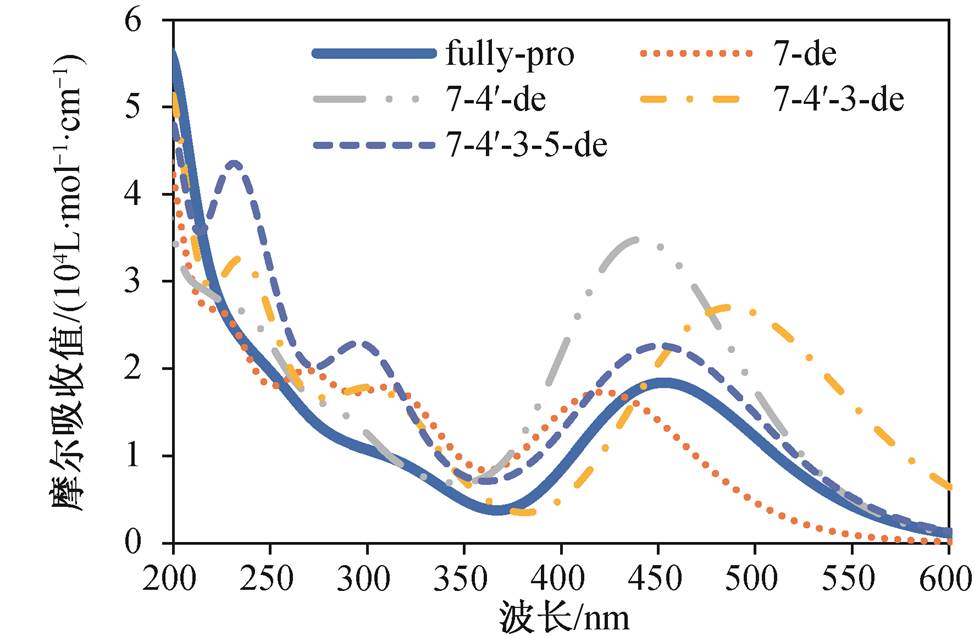
图3 5种去质子化结构的紫外–可见光计算光谱
Fig. 3 Calculated UV-vis spectra of five deprotonated structures
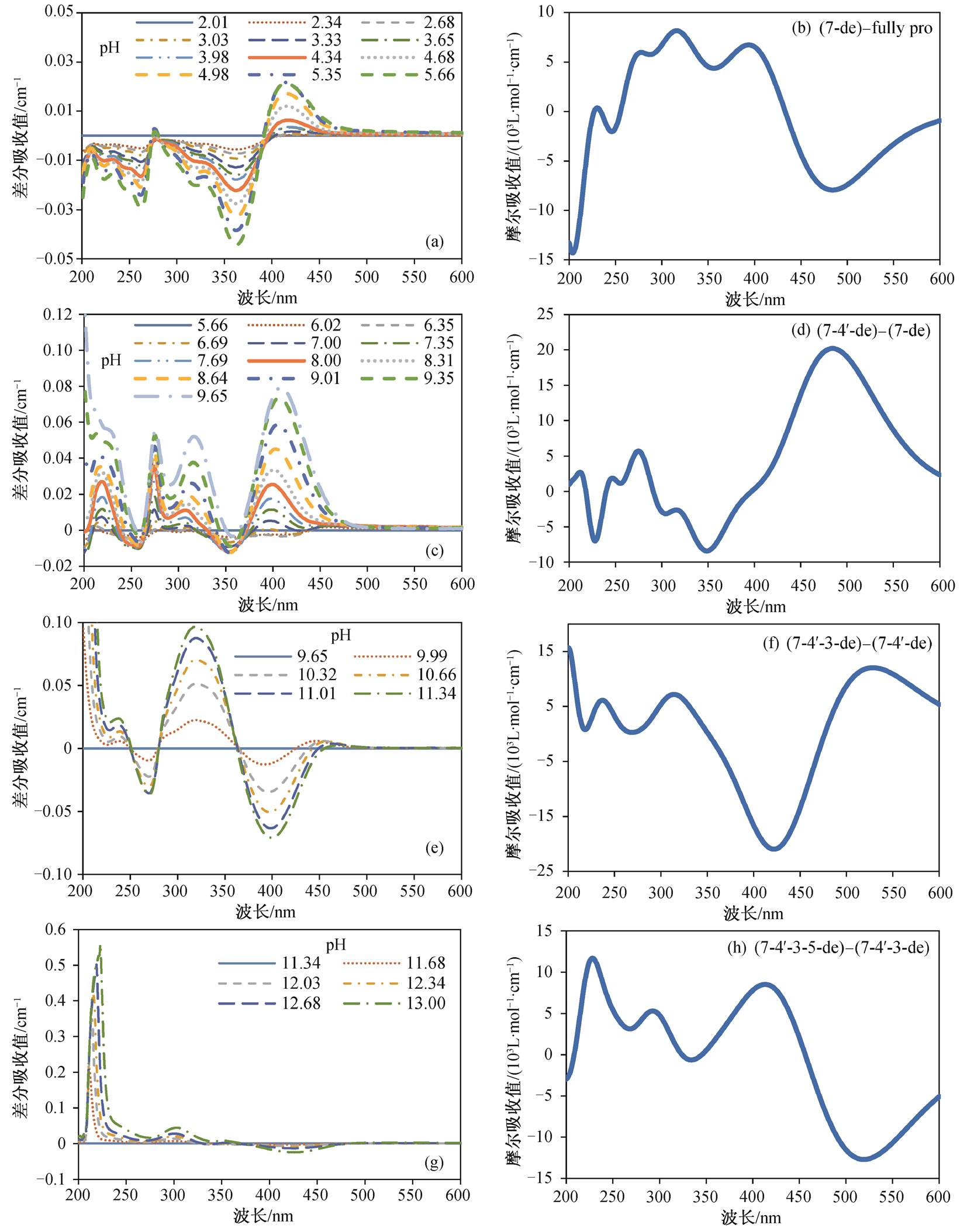
(a)以pH 2.01作差的实验差分光谱; (b)以完全质子化结构作差的7号去质子化结构计算差分光谱; (c)以pH 5.66作差的实验差分光谱; (d)以7号去质子化结构作差的7-4′号去质子化结构计算差分光谱; (e)以pH 9.65作差的实验差分光谱; (f)以7-4′号去质子化结构作差的7-4′-3号去质子化结构计算差分光谱; (g)以pH 11.34作差的实验差分光谱; (h)以7-4′-3号去质子化结构作差的7-4′-3-5号去质子化结构计算差分光谱
图4 山奈酚去质子化过程的实验光谱和计算光谱对比
Fig. 4 Comparison of experimental and calculated spectra of ferulic acid induced by its deprotonation
整体而言, 计算值与实验值吻合得很好, 进一步证实山奈酚去质子化过程是7, 4′, 3, 5号酚基依次去质子化。其中, 200~400nm区间内量子化学计算结果与实验值吻合度较高, 一些计算值甚至与实验值完全一致。然而, 在400nm以上区间, 计算结果与实验结果差距较大。
从量子化学角度分析, 分子的性质与其电子分布密切相关, 分子轨道反映电子在整个分子内的运动状态, 其中最重要的分子轨道为最高占据分子轨道(highest occupied molecular orbital, HOMO)和最低未占据分子轨道(lowest unoccupied molecular orbi-tal, LUMO), HOMO与LUMO是分子轨道跃迁最低能量差, 因此HOMO表示最容易失去电子的占据轨道, LUMO表示最容易得到电子的未占据轨道。
根据前线轨道理论(frontier molecular orbital theory, FMO), 去质子化过程的差分吸收光谱中峰的出现与HOMO/LUMO附近的分子轨道(HOMO-1, HOMO-2等或LUMO+1, LUMO+2等, HOMO-1代表次高占据分子轨道, LUMO+1代表次低未占据分子轨道, 依次类推)之间的跃迁过程紧密相关 [14–16] 。此过程同时伴随去质子化前、后分子内电子激发引起的电子分布变化。电子激发是从 n 个分子轨道到其他 n 个分子轨道之间的跃迁, 每个分子轨道都可能涉及, 这可由TD-DFT计算得以清晰的阐释。通过计算可知, 山奈酚7号酚基去质子化结构计算光谱中278 nm峰对应的分子轨道跃迁为HOMO到LUMO (图5)。分子轨道用两种颜色(红色和绿色)显示, 分别代表分子轨道波函数两种不同相位(也称为波函数的正负)。图 5 中覆盖红色或绿色的区域代表此处电子云出现的概率, 即电子密度。某处电子密度越大, 则颜色响应区域越大。
山奈酚与Cu 2+ 络合滴定实验光谱如图6所示。根据Cu 2+ 浓度递增顺序, 能够发现吸收光谱在不同波长区间内的上升或下降。与解离过程同理, 通过作差处理得到两段不同Cu 2+ 浓度区间的差分吸收光谱, 分别为低浓度区间和高浓度区间, 两个阶段具有各自的光谱变化特征。
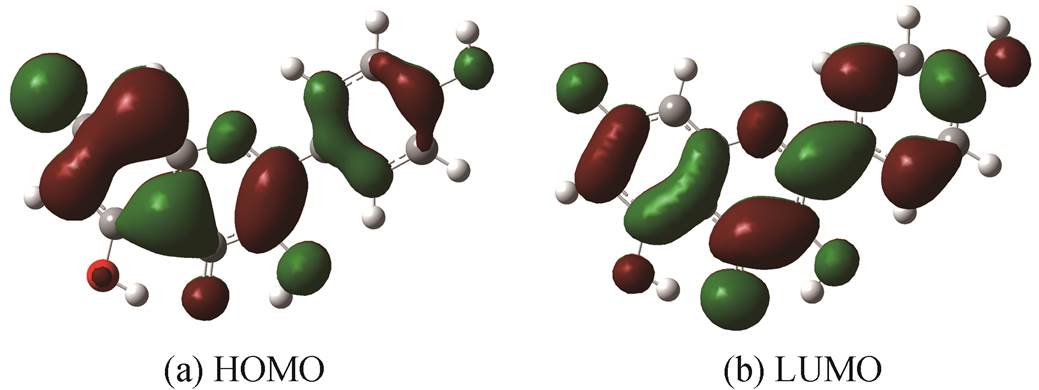
红色和绿色区域分别表示波函数的正值和负值, 白色和灰色小球分别代表氢原子和碳原子, 下同
图 5 7号酚基去质子化结构计算光谱中278nm峰对应的分子轨道
Fig.5 Molecular orbitalscorrespondingtothepeakat278nm in the calculated spectra in 7-deprotonation form
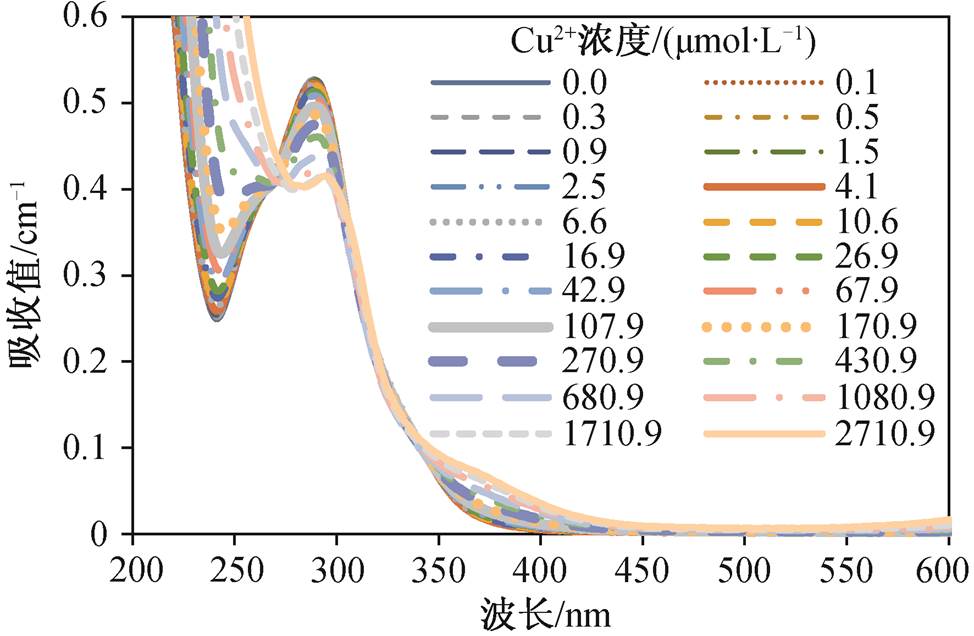
图6 不同Cu 2+ 浓度时山奈酚的吸收光谱
Fig. 6 Absorbance spectra of kaempferol at various concentrations of Cu(II)
为了研究山奈酚与Cu 2+ 络合的差分光谱中峰的产生机理, 需要计算可能的络合结构。山奈酚分子含4个羟基, 它们是发生络合效应的主要基团。由于4号酮基处于3号和5号酚基的中间位置, 氧原子带负电, 因此O会与酚基共同参与Cu 2+ 的络合, 形成三角形稳定结构。另外, 当酚基与Cu 2+ 络合时, 由于酚基上的氢原子带正电荷, 会被Cu 2+ 的排斥作用脱去 [17–18] 。基于上述分析, 在量子计算时, 对山奈酚-Cu 2+ 络合结构做出如下模型的搭建: A. Cu同时与3号、4号络合(Cu-3-4); B. Cu同时与4号、5 号络合(Cu-4-5); C. Cu只与3号络合(Cu-3); D. Cu只与4号络合(Cu-4); E. Cu只与5号络合(Cu-5); F. Cu只与7号络合(Cu-7); G. Cu只与4′号络合(Cu-4′)。其中, A和B为Cu 2+ 与两个氧原子同时络合, C, D, E, F和G为Cu 2+ 与一个氧原子络合。
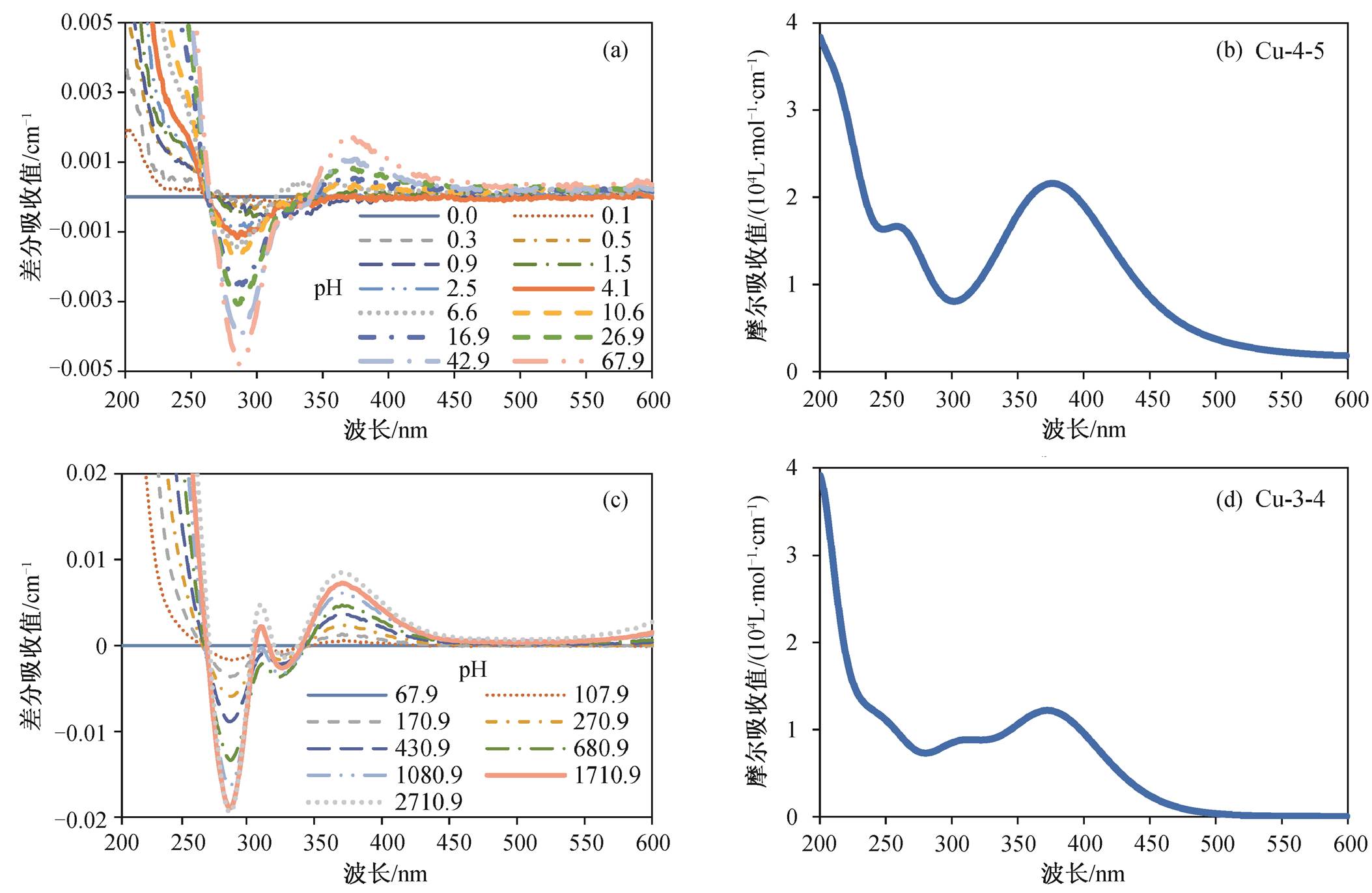
(a) 67.9 μmol/L 以下Cu 2+ 浓度的实验差分光谱; (b) Cu-4-5络合结构的计算光谱; (c) 67.9~2710.9 μmol/L Cu2+浓度的实验差分光谱;(d) Cu-3-4络合结构的计算光谱
图7 山奈酚与Cu 2+ 络合过程的实验光谱和计算光谱对比
Fig. 7 Comparison of experimental and calculated spectra of kaempferol and its complexes with Cu(II)
山奈酚与Cu 2+ 络合过程的两段实验光谱和计算光谱对比如图7所示。在0~67.9μmol/L Cu 2+ 浓度区间内, 山奈酚的差分实验光谱显示在250和373nm处出峰, 计算光谱中出现的258和376nm分别与实验值非常接近。在67.9~2710.9μmol/LCu 2+ 浓度区间内, 山奈酚的差分实验光谱显示在311和368nm处出峰, 计算差分光谱中出现的312和372nm分别与实验值非常接近。
通过比较可以看出, 计算值与实验值能够很好吻合, 说明山奈酚与Cu 2+ 的络合过程是低浓度下Cu 2+ 同时与4号和5号酚基络合, 高浓度下Cu 2+ 同时与3号和4号酚基络合。
与去质子化过程同理, 络合过程的差分吸收光谱中峰的出现与HOMO/LUMO附近的分子轨道之间的跃迁过程紧密相关, 此过程同时伴随络合前、后分子内电子激发引起的电子分布变化, 可由TD-DFT计算清晰地得以阐释。通过计算可知, Cu 2+ 与山奈酚4号、5号酚基络合结构计算光谱中376 nm峰对应的分子轨道跃迁为HOMO-1到LUMO, 分子轨道如图 8 所示。
本文结合紫外–可见光差分吸收光谱和量子化学计算两种方法, 对山奈酚的解离(去质子化)过程及与Cu 2+ 络合过程进行研究, 得到以下主要结论。
1)紫外可见光光谱法灵敏度高, 其差分吸收光谱(DAS)能够精细地表征解离实验中pH=0.33和金属络合过程1μmol/L以内的变化, 是研究复杂水化学过程的有力工具。
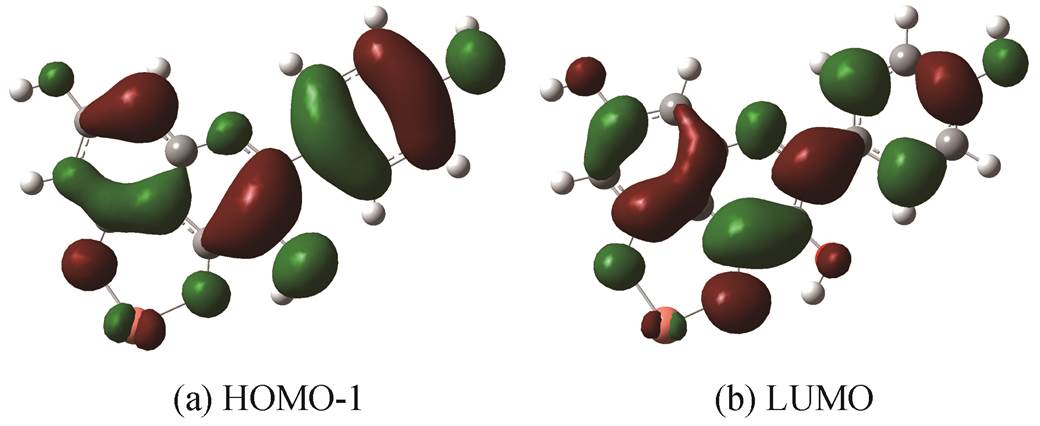
图 8 Cu 2+ 与 4 号、5 号酚基络合结构计算光谱中 376nm峰对应的分子轨道
Fig.8 Molecular orbitals corresponding to the peak at 376 nm in the calculated spectra in Cu-4-5 binding form
2)山奈酚在pH从2.01增至13.00过程中的吸收光谱变化较复杂。通过量子化学计算各种可能去质子化结构的Gibbs能量, 发现解离过程是按7, 4′, 3, 5号酚基的顺序依次去质子化。通过依次比较4个阶段的差分光谱与计算差分光谱, 证实了该去质子化顺序。计算光谱与差分光谱能够很好地吻合。
3)山奈酚与Cu 2+ 络合过程的吸收光谱较有规律。通过量子化学计算各种可能络合结构的UV-vis光谱, 发现山奈酚与Cu 2+ 的微观络合过程: 在低浓度(<67.9 μmol/L)Cu 2+ 时, 4号和5号酚基同时与Cu 2+ 络合; 在高浓度(67.9~2710.9 μmol/L)Cu 2+ 时, 3号和4号酚基同时与Cu 2+ 络合。计算光谱与差分光谱能够很好地吻合。
4)山奈酚的解离以及与Cu 2+ 络合过程中, 特征峰的出现是由解离、络合前后活性基团电子分布的变化及光子照射时HOMO到LUMO能量吸收的变化导致, 从而揭示了山奈酚解离及金属络合过程的机理。
参考文献
[1]Calderon-Montano M J, Burgos-Morón E, Pérez-Guerrero C, et al. A review on the dietary flavonoid kaempferol. Mini Reviews in Medicinal Chemistry, 2011, 11(4): 298–344
[2]Liu R H. Health-promoting components of fruits and vegetables in the diet. Advances in Nutrition: An International Review Journal, 2013, 4(3): 384S–392S
[3]Kim S H, Choi K C. Anti-cancer effect and underlying mechanism(s) of kaempferol, a phytoestrogen, on the regulation of apoptosis in diverse cancer cell models. Toxicol Res, 2013, 29(4): 229–234
[4]Perron N R, Brumaghim J L. A review of the antio- xidant mechanisms of polyphenol compounds related to iron binding. Cell Biochemistry and Biophysics, 2009, 53(2): 75–100
[5]Yan M, Dryer D, Korshin G V, et al. In situ study of binding of copper by fulvic acid: comparison of differential absorbance data and model predictions. Water Research, 2013, 47(2): 588–596
[6]Yan M, Korshin G V. Comparative examination of effects of binding of different metals on chromo- phores of dissolved organic matter. Environmental Science & Technology, 2014, 48(6): 3177–3185
[7]Frisch M J, Trucks G W, Schlegel H B, et al. Gaussian 2009 (Revision D. 1). Pittsburgh: Gaussian Inc, 2009
[8]Runge E, Gross E K U. Density-functional theory for time-dependent systems. Physical Review Letters, 1984, 52(12): 997 - 1000
[9]Becke A D. Density-functional thermochemistry. III. The role of exact exchange. The Journal of Chemical Physics, 1993, 98(7): 5648–5652
[10]Lee C, Yang W, Parr R G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Physical Review B, 1988, 37 (2): 785 - 789
[11]Hay P J. Gaussian basis sets for molecular calcula- tions. The representation of 3 d orbitals in transition-metal atoms. The Journal of Chemical Physics, 1977, 66(10): 4377–4384
[12]Ren J, Meng S, Lekka C E, et al. Complexation of flavonoids with iron: structure and optical signatures. The Journal of Physical Chemistry B, 2008, 112(6): 1845–1850
[13]Tsolakidis A, Kaxiras E. A TDDFT study of the optical response of DNA bases, base pairs, and their tautomers in the gas phase. The Journal of Physical Chemistry A, 2005, 109(10): 2373–2380
[14]Cornard J P, Dangleterre L, Lapouge C. DFT and TD-DFT investigation and spectroscopic characterization of the molecular and electronic structure of the Zn(II)-3-hydroxyflavone complex. Chemical Physics Letters, 2006, 419(1): 304–308
[15]Say-Liang-Fat S, Cornard J P. Al(III) complexation by alizarin studied by electronic spectroscopy and quan- tum chemical calculations. Polyhedron, 2011, 30(13): 2326–2332
[16]Lapouge C, Dangleterre L, Cornard J P. Spectroscopic and theoretical studies of the Zn(II) chelation with hydroxyflavones. The Journal of Physical Chemistry A, 2006, 110(45): 12494–12500
[17]Le Person A, Moncomble A, Cornard J P. The com- plexation of A lIII , Pb II , and Cu II metal ions by esculetin: a spectroscopic and theoretical approach. The Journal of Physical Chemistry A, 2014, 118(14): 2646–2655
[18]Moncomble A, Cornard J P. Elucidation of comple- xation multi-equilibrium with Mg II and a multisite ligand. A combined electronic spectroscopies and DFT investigation. RSC Advances, 2014, 4(55): 29050–29061
Revealing the Mechanism of Hydrolysis and Metal Complexation of Kaempferol by Differential Absorbance Spectra and TD-DFT Calculations
HAN Xuze, YAN Mingquan †
The Key Laboratory of Water and Sediment Sciences (MOE), Department of Environmental Engineering, Peking University, Beijing 100871
Abstract In order to reveal the mechanism of hydrolysis and metal complexation of kaempferol, the UV-vis spectra of kaempferol in hydrolysis process (pH from 2.01 to 13.00) and in Cu(II) complexation process (from 0.1 to 2710.9 μmol/L) was obtained, and the characteristic spectra at various reaction stages was obtained by differ-ential treatment. Combined with quantum chemical theoretical computation, a variety of possible deprotonation structures, complexation structures and corresponding electron absorption spectra were calculated based on DFT (Density Functional Theory) and TD-DFT (Time-Dependent Density Functional Theory). The experimental results are compared with the calculated values and it indicates that the hydrolysis process is in the deprotonation order of 7, 4′, 3, 5 phenolic group; at low concentrations of Cu(II) (<67.9 μmol/L), Cu(II) chelated with 4,5 phenolic group simultaneously; at high concentration of Cu(II) (67.9–2710.9 μmol/L), Cu(II) chelated with 3,4 phenolic group simultaneously. The calculated spectra is consistent with experimental spectra in hydrolysis and Cu(II) com-plexation process of kaempferol. The appearance of characteristic peaks is due to the change of the electron distribution of the active groups before and after hydrolysis and Cu(II) complexation, resulting in the changes of energy gap from HOMO (highest occupied molecular orbital) to LUMO (lowest unoccupied molecular orbital) during photon irradiation. The mechanism of hydrolysis and metal complexation process of kaempferol was revealed.
Key words differential absorbance spectra; quantum chemical computation; hydrolysis; complexation; molecular orbital
doi: 10.13209/j.0479-8023.2017.117
中图分类号 X142
收稿日期: 2017-05-19;
修回日期: 2017-05-29;
网络出版日期: 2017-07-05
†通信作者 , E-mail: yanmq@pku.edu.cn
† Corresponding author , E-mail: yanmq@pku.edu.cn
国家自然科学基金(51578007)资助